Gabriela Silvia Gheorghe1, Andreea Simona Hodorogea1, Ioan Tiberiu Nanea1
1 University of Medicine and Pharmacy ”Carol Davila”, Faculty of Medicine, Bucharest, Romania, ”Theodor Burghele” Clinical Hospital
Contact address:
Gabriela Silvia Gheorghe, Adress: ”Theodor Burghele” Clinical Hospital, No 20 Panduri Road, Bucharest, Romania, postal code 050653.
E-mail: gsgheorghe@yahoo.com.
Abstract: Statins are probably the most prescribed drugs in cardiology worldwide, with evident efficiency in primary and secondary prevention. They act on the first step of cholesterol synthesis by blocking the hepatic hydoxi methyl glutaril coenzyme A (HMG CoA) reductase dose-dependent, reversible and competitive. Consequently, not only the cholesterol synthesis is blocked, but also the synthesis of many other intermediate products involved in other important biological processes: mythocondrial respiratory chain, inflammation, oncogenesis. These changes can explain the non lipidic (pleiotrophic) effects of statins, but also their adverse effects. The pharmacology of statins is complex. They differ in terms of lipophilicity, active or inactive form of administration, active metabolites, elimination. There are genes correlated with the susceptibility of patients to adverse effects. Despite many studies, there is few data regarding the clinical implications of the pharmacological peculiarities of statins. This paper discusses some clinical issues about the biochemistry and pharmacology of statins.
Keywords: statins; HMG Co A reductase; isoprenoids; farnesyl pirophosphate; geranyl pirophosphate; ubiquinone
Rezumat: Statinele sunt printre cele mai prescrise medicamente în cardiologie, cu eficiență dovedită în prevenţia primară şi secundară a bolilor cardiovasculare. Ele acționează în prima etapă a sintezei hepatice de colesterol prin inhibarea competitivă, dependentă de doză și reversibilă a hidoxi metil glutaril coenzimei A (HMG CoA). Ca urmare, este blocată nu doar sinteza colesterolului, dar și cea a multor produși intermediari implicați în procese biologice importante: lanțul respirator mitocondrial, inflamație, oncogeneză. Aceste modificări ar putea explica efectele nonlipidice (pleiotrope) ale statinelor, dar și efectele adverse ale acestora. Statinele au o farmacologie complexă, fiind diferite între ele din punct de vedere al liposolubilității, al formei de administrare – activă sau inactivă, al metaboliților activi, al formei de eliminare. Susceptibilitatea la efectele adverse se corelează cu prezența anumitor gene. Deși studiile în care s-au folosit statine sunt foarte numeroase, efectele determinate de particularitățile farmacologice ale acestora nu sunt bine cunoscute. Articolul se referă la o serie de aspecte clinice în relație cu biochimia și farmacologia statinelor.
Cuvinte cheie: statine, HMG Co A reductaza, izoprenoizi, farnezil pirofosfat, geranil pirofosfate, ubichinona
Introduction
Statins are among the most studied and prescribed drugs, indicated in primary and secondary prevention of cardiovascular diseases. They are structural analogs of mevalonate and act on the rate limiting first step of the cholesterol synthesis by blocking the hepatic hydoxi methyl glutaril coenzyme A (HMG CoA) reductase1. Given their importance in clinical practice, but also the existence of less well-known data, we conducted a review of the latest data from the medical literature using PubMed search through internet. The keywords used were „statins”, „HMG Co A reductase”, „isoprenoids”, „farnesyl pirophosphate”, „geranyl pirophosphate”, „ubiquinone”. The research lasted five months.
History
Statin history began in 1971, when the Japanese biochemist Akira Endo (Sankyo) discovered microorganisms that synthesize inhibitors of HMG Co A reductase as a defense mechanism, since mevalonate is a precursor of cell wall components (ergosterol) and of the cytoskeleton (isoprenoids)2. He identified Mevastatin produced by the fungus Penicillium citrum. In 1976 British researchers isolated Mevastatin (Compactin) of the fungus Penicillium brevicompactum, never marketed because of adverse effects. In 1978 P. Roy Vagelos (Merck&Co) isolated Lovastatin of the fungus Aspergillus cereus, the first statin sold as lipid lowering agent, under the name Mevacor (1987). Soon appeared Pravastatin, produced by fermentation of Nocardia autotrophica (1988), Simvastatin, synthetically derived from the products of the fermentation of Aspergillus terreus, Fluvastatin, Atorvastatin, Rosuvastatin and Pitavastatin, the last 3 entirely produced by synthesis. Cerivastatin, very strong lipid-lowering statin also produced by synthesis, was withdrawn from the market because of adverse effects.
Cholesterol synthesis
Cholesterol synthesis is a complex process (Figure 1) strictly regulated both short and long term (Figure 2). The molecules of geranyl and farnesyl occurring in intermediate stages of cholesterol synthesis bind various prenylated proteins to cell membranes. Among these proteins there are some with cellular signals role, such as those related to Guanosine 5’-Monophosphate (GMP) system and Ras gene, involved in oncogenesis. Another isoprenoid is dolichol in cell membranes, involved in the synthesis of carbohydrate chains of glycoproteins. Coenzyme Q (ubiquinone), with a role in electron transfer in the mithocondrial respiratory chain, includes an isoprenoid side chain. Hem also has a farnesyl side chain.
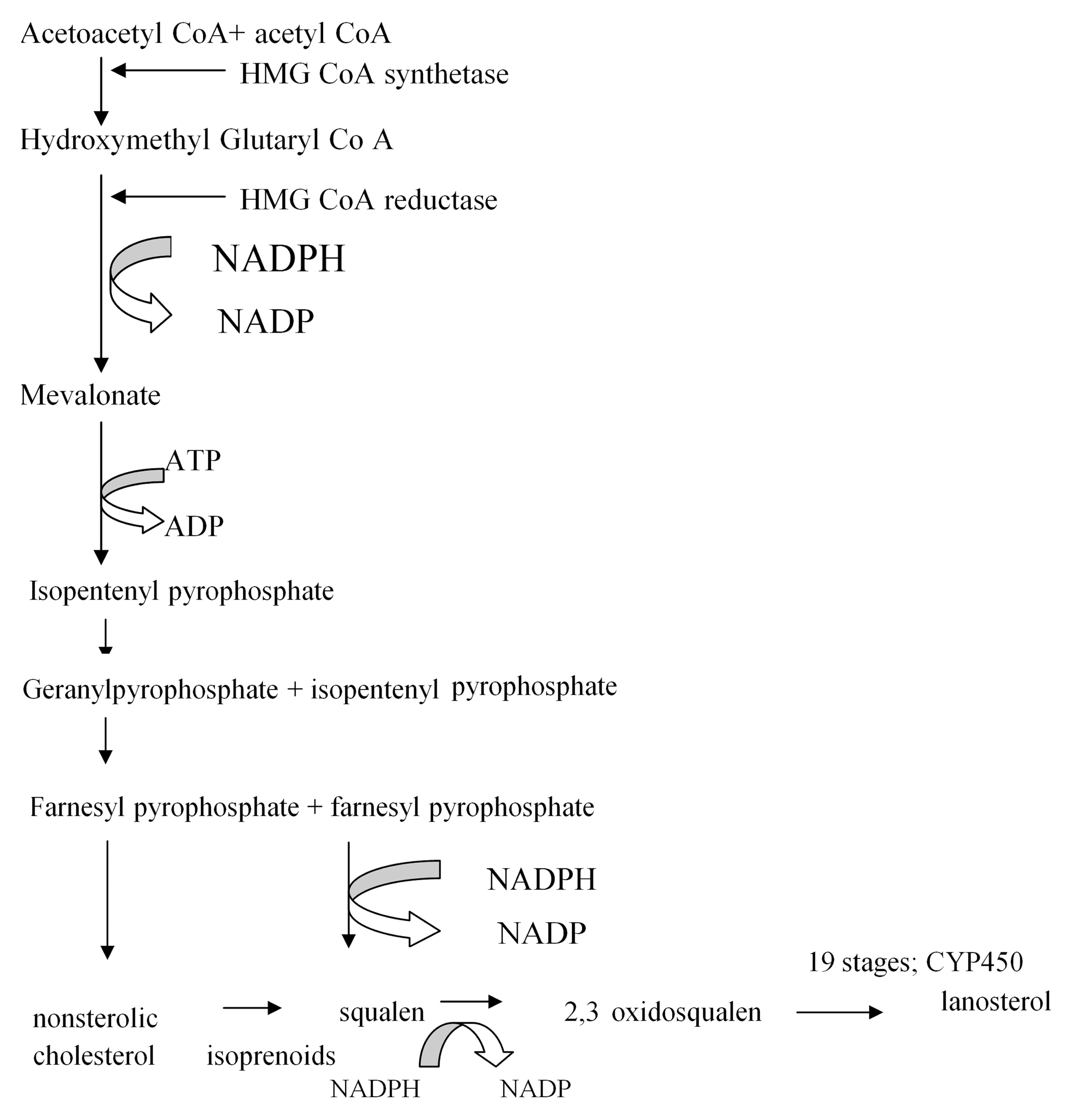
Figure 1. Hepatic cholesterol synthesis.
Hepatic cholesterol synthesis is an energy consuming process, involving cytochrome P450 (CYP450). It starts by condensing acetoacetyl coenzyme A (CoA) to acetyl CoA by the action of cytoplasmic hidroximetilglutaril CoA synthase (HMG-CoA). CoA – coenzyme A, NADP – nicotinamide adenine dinucleotide phosphate, NADPH – he reduced form of nicotinamide adenine dinucleotide phosphate, ATP – adenosine triphosphate, ADP – adenosine diphosphate
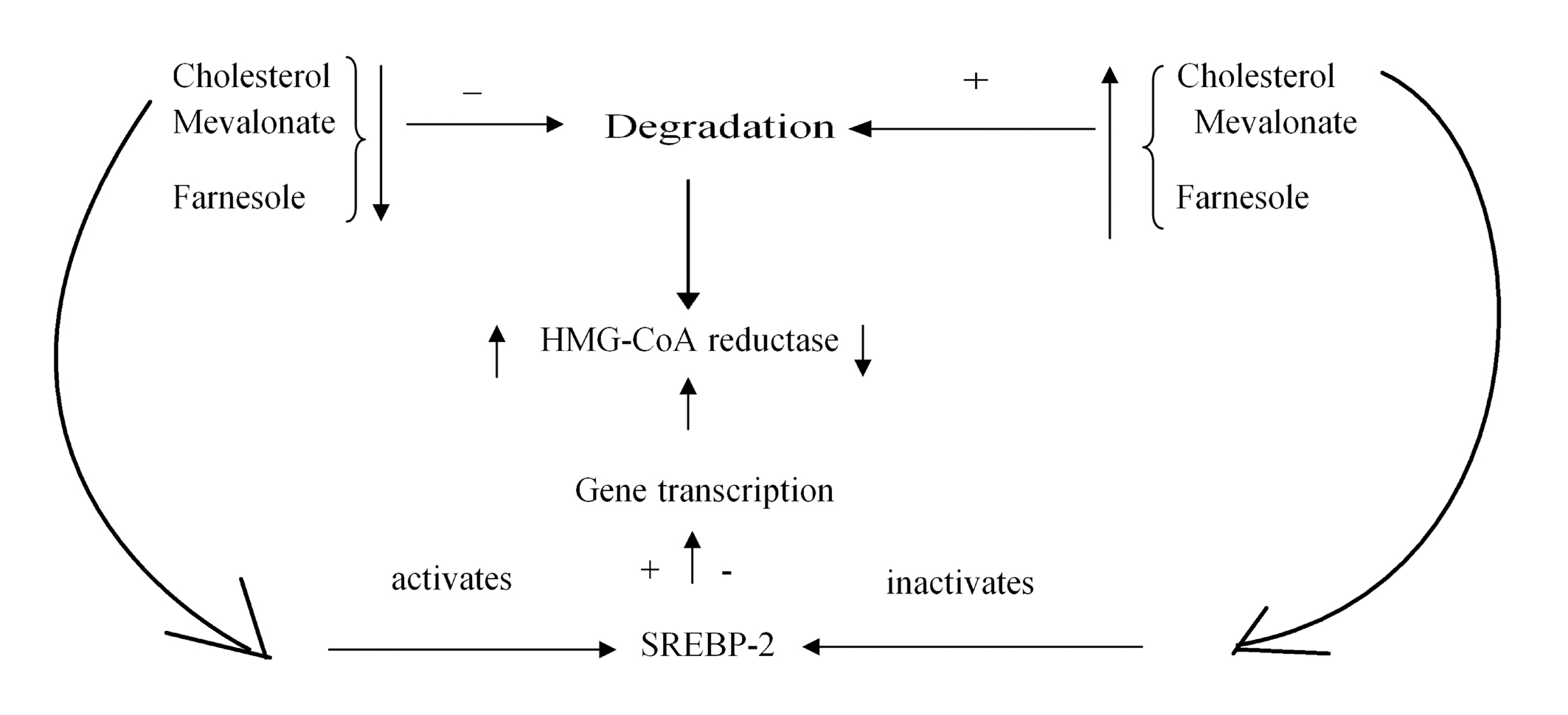
Figure 2. Long term adjustment of cholesterol synthesis.
Reducing the amount of cholesterol, mevalonate, farnesole inhibits the degradation of the hydoxi methyl glutaril coenzyme A (HMG CoA) reductase. Reducing cholesterol activates SREBP-2 (sterol regulatory element binding protein) in the membrane of the endoplasmic reticulum, which migrates into the nucleus and induces transcription, and thus synthesis of HMG-CoA reductase. Increasing the amount of cholesterol has reverse effects.
Short-term control of cholesterol synthesis is achieved by the intervention of an adenosine monophosphate (AMP)-dependent protein kinase, whose activity depends on cellular energy levels. Increasing the amount of intracellular AMP (thus reducing the amount of adenosine triphosphate (ATP)) activates the protein kinase that phosphorylates and inhibits HMG-CoA reductase1.
Long term control of cholesterol synthesis is made by adjustment of the HMG-CoA reductase synthesis and degradation. The synthesis of HMG-CoA reductase increases by its transcription activation by the action of SREBP-2 (Sterol Regulatory element binding proteins) in the endoplasmic reticulum membrane. Reducing the amount of cholesterol leads through complex mechanisms, to the cleavage between the SREBP-2 and the protein that keeps it anchored to the endoplasmic reticulum membrane. Thus, SREBP-2 migrates to the nucleus and increases the transcription of the HMG-CoA reductase gene (Figure 2)1.
Pharmacology of statins
The chemical structure of statins
Statins consist of two components: the pharmacophore, dihydroxyheptanoic acid derivative, that inhibits HMG-CoA reductase competitive, reversible, dose-dependent; a peripheral ring differently substituted, covalently bound to the pharmacophore1. It binds statin to HMG-CoA reductase, preventing its removal by endogenous substrate, HMC-CoA. For each statin the substituents attached to the ring are different and they determine its solubility3,4 (Figure 3). Lovastatin, fluvastatin, simvastatin, atorvastatia, pitavastatin are lipophilic and pravastatin and rosuvastatin are hydrophilic. Statins lipophilicity influence their systemic bioavailability.
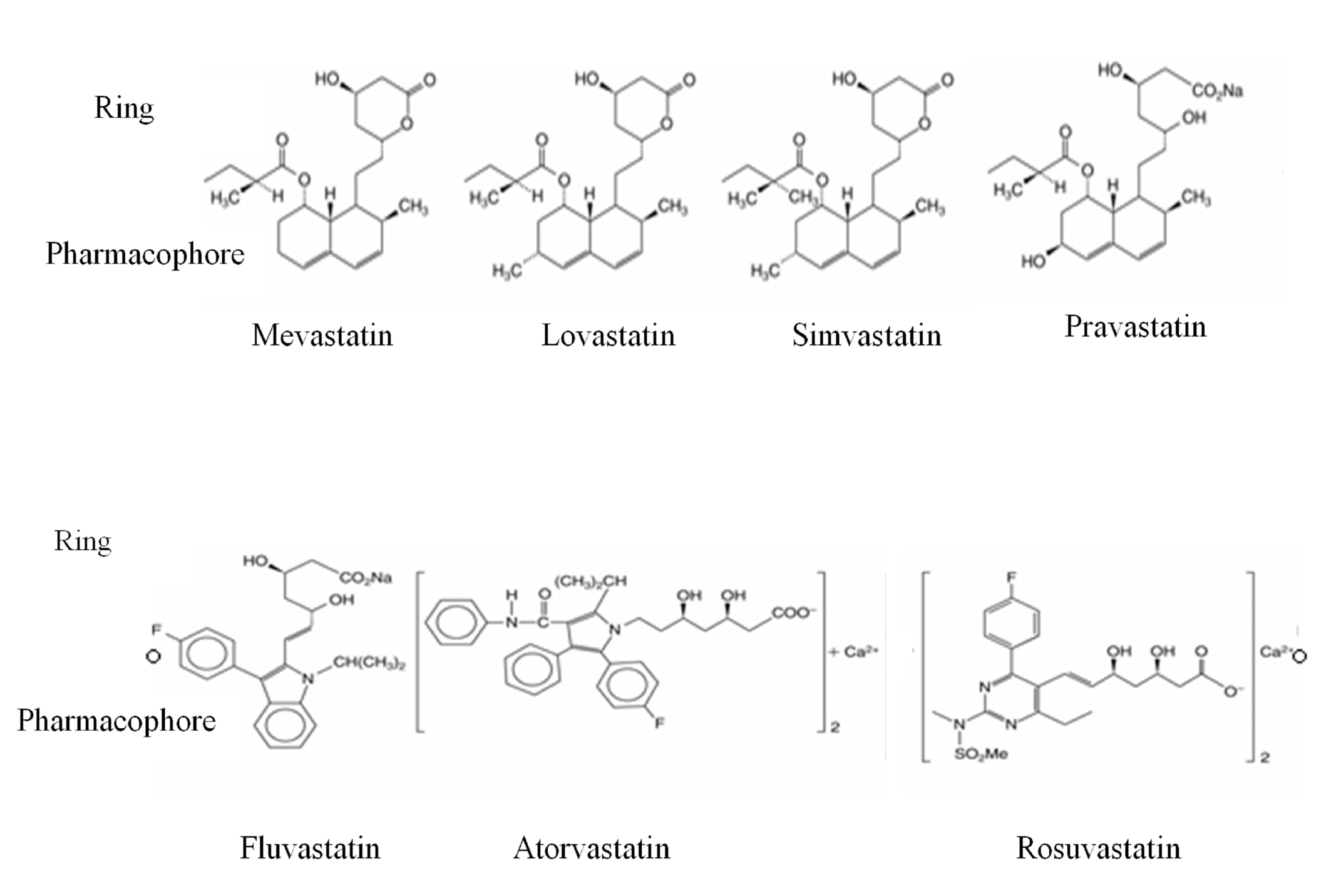
Figure 3. Chemical structure of statins4.
Pharmacokinetics
Statins are given orally, most of them in the active form of the hydroxy acid. Lovastatin and simvastatin are given in inactive form, as lactone3, more lipophilic than hydroxy acids5. They are absorbed in the intestinal cells by passive mechanism and by active transport, using transporters synthesized by ABC and SCL gene families. Prodrug activation begins in the intestinal mucosa. Their absorption varies between 30% (lovastatin) and 98% (fluvastin)6. Lovastatin is absorbed better when administered once with food; fluvastatin, atorvastatin, pravastatin are better absorbed by fasting and simvastatin and rosuvastatin absorption is not influenced by food. Absorption is fast and the maximum level is reached in about 4 hours. The blood concentration is not correlated with the intensity of effects3. Plasma protein binding is over 95% for fluvastatin, lovastatin, simvastatin, atorvastatin and 35-50% for pravastatin and rosuvastatin5,7. Lipophilic statins enter the hepatocyte by passive diffusion. Partly they remain in circulation and enter the peripheral tissues by passive mechanism due to their liposolubility. Hydrophilic statins are captured by hepatocytes using specific carriers that do not exist in extrahepatic tissues. Their extrahepatic distribution is reduced. Statins administered as lactones are activated in the liver to the acid form (beta hydroxy acids) by intracellular hydrolysis using esterases and paroxonases. Intrahepatic metabolism of statins is complex, involving reversible transformation between the lactone form (fat soluble) and acid form (less fat soluble), CoA, UDP glucuronosyl transferase glucuronidation and microsomal cytochrome P450 (CYP) participation8. Simvastatin, atorvastatin, lovastatin are metabolised by CYP 3A4 pathway to active metabolites. Fluvastatin is metabolized predominantly by CYP2C9 pathway, but also by CYP3A4 and CYP2C8 to mostly inactive metabolites3. Rosuvastatin is minimum inactivated by CYP2C9 and CYP2C19 pathway. Pravastatin is partially degraded in the stomach and limited metabolized in the cytosol of hepatocytes, by CYP 450-independent pathways5. Pitavastina is limited metabolized predominantly by glucuronidation3. Lipophilic statins are more likely to be metabolised by CYP 4505,6. Pravastatin, rosuvastatin, pitavastatin are excreted almost unchanged5. Statins are active eliminated predominantly by hepatic biliary excretion, with P-glycoprotein and MRAP-2 (Multidrug Resistance Protein Associated 2) transporters participation in the basal-lateral membrane of hepatocytes. Renal elimination is reduced, except for pravastatin, 60% eliminated by renal tubular secretion. Rosuvastatin is removed by the kidneys and the liver, mostly unchanged4. The half-life is 0.5-3 hours, with the exception of atorvastatin (10 hours) and rosuvastatin (12 hours). It is recommended to take statins in the evening because cholesterol synthesis is more active at night. Atorvastatin and rosuvastatin may be given at any of the day5 (Table 2).
Mechanism of action
Statins reversible inhibit microsomal HMG-CoA reductase and decrease intracellular cholesterol biosynthesis. As a result, there is the phenomenon of transcriptional up regulation of the production of microsomal HMG-CoA reductase and also of the number of membrane receptors for LDL cholesterol. The circulating LDL cholesterol particles are trapped intracellular and resets cholesterol homeostasis in extrahepatic tissues, but does not markedly influence global cholesterol balance9. The LDL cholesterol level in blood decreases in 4-6 weeks, usually by 20-35%, depending on the statin dose and not on the statin blood concentration. This is due to the complex metabolism of statins, during which active products may appear. The lipid-lowering effect of statins depends on the genetic polymorphism of HMG-CoA reductase and there are patients with low response3.
Clinical studies
The widespread use of statins coincided with the understanding of the role of cholesterol in atherosclerosis and with the development of primary and secondary prevention in ischemic heart disease. If in 1984 there were few prevention studies, the best known being The Coronary Primary Prevention Trial (cholestyramine)8, there are now more than 70 primary and secondary prevention studies, involving over 170,000 subjects7 (Table 1).
Table 1. Primary and secondary prevention trials with statins in atherosclerotic disease
| Trial/Primary prevention | Statin/ mg | Versus | No subjects | Duration (years) |
| AFCAPS/TEXCAPS (1998) | lovastatin/20-40 | placebo | 6605 | 5.2 |
| EXCEL (1991) | lovastatin/20-80 | placebo | 8245 | 0.9 |
| WOSCOPS (1995) | pravastatin/40 | placebo | 6595 | 4.9 |
| PROSPER (2002) | pravastatin/40 | placebo | 5800 | 3.2 |
| ALLHAT –LLT (2002) | pravastatin /40 | placebo | 10355 | 4.8 |
| HPS (2002) | simvastatin/40 | placebo | 20536 | 5 |
| ASAP (2001) | atorvastatin/40-80 | simvastatin/20-40 | 325 | 2 |
| ASCOT-LLA (2004) | atorvastatin/10 | placebo | 10305 | 3.3 |
| CARDS (2004) | atorvastatin/10 | placebo | 2838 | 4 |
| ASTEROID (2008) | rosuvastatin 40 | opened | 292 | 2 |
| JUPITER (2008) | rosuvastatin/20 | placebo | 17802 | 4 |
| METEOR (2007) | rosuvastatin 40 | placebo | 984 | 2 |
| Trial/Secondary prevention | statin | versus | No pts/years | Duration (years) |
| LIPS (2002) | fluvastatin/80 | placebo | 1677 | 3.9 |
| LIPID (1998) | pravastatin/40 | placebo | 9014 | 6.1 |
| PROVE-IT-TIMI22 (2004) | pravastatin 40 | atorvastatin 80 | 4162 | 2 |
| CARE (1996) | pravastatin/40 | placebo | 4159 | 5 |
| GISSI-P statin (2000) | pravastatin/20 | usual treatment | 4271 | 2 |
| 4S (1994) | simvastatin/20 | placebo | 4444 | 5.4 |
| SCAT (2000) | simvastatin/10-40 | placebo | 460 | 4 |
| A to Z (2004) | simvastatin/40-80 | placebo | 2265 | 1-4 luni |
| SEARCH (2010) | simvastatin 20 | simvastatin 80 | 12064 | 6.7 |
| TNT (2005) | atorvastatin 80 | atorvastatin 10 | 15 464 | 4.9 |
| ALLIANCE (2004) | atorvastatin/40-80 | usual treatment | 2442 | 4.3 |
| IDEAL (2006) | atorvastatin 80 | simvastatin 40 | 8888 | 4.8 |
| MIRACLE (2001) | atorvastatin/80 | placebo | 3086 | 0.33 |
| GREACE (2002) | atorvastatin/10-80 | usual treatment | 166 | 3 |
| CORONA (2007) | rosuvastatin/10 | placebo | 5011 | 3 |
| GISSI-HF85 (2008) | rosuvastatin/10 | placebo | 4574 | 3.9 |
| A to Z (Aggrastat to Zocor); LIPS (Lescol Interventional Prevention Study); LIPID (The Long-Term Intervention with Pravastatin in Ischaemic Disease ); TNT (Treating to New Target) PROVE IT TIMI 22 (A Comparison of Intensive Statin Therapy and Moderate Statin Therapy in Acute Coronary Syndrome Patients). MIRACLE (Myocardial Ischhemia Reductiion and Aggressive Cholesterol Lowering Trial); SEARCH (Intensive lowering of LDL cholesterol with 80 mg versus 20 mg Simvastatindaily in 12064 survivors of myocardial infarction: a doublee blind randomized study) 4S (Scandian Survival Simvastatin Study) IDEAL (Incremental Decrease in Endpoints through Aggressive Lipid Lowering Study); GISSI-HF85 (Effects of Rosuvastatin in Chronic Heart Failure; a randomized, placebo control double blind trial) CORONA (Control Rosuvastatin in Multinational Trial in Heart Failure); GREACE (Safety and efficacy of long-term statin treatment for cardiovascular events in patients with coronary heart disease and abnormal liver tests in the Greek Atorvastatin and Coronary Heart Disease Evaluation ) ALLIANCE (Aggressive Lipid Lowering versus Usual Care in a Managed-Care Patient Population) | ||||
Reduction of LDL cholesterol by 20-35% is associated with 30-40% reduction in major cardiovascular events9. This decrease is achieved with different doses of different statins: atorvastatin 10 mg, rosuvastatin 5 mg, simvastatin 20 mg, fluvastatin 80 mg, lovastatin 40 mg, pravastatin 40 mg. Doubling statin dose leads to further reduction of LDL cholesterol level by 6%9,10. If there is a need to decrease LDL cholesterol by 50% from baseline, atorvastatin 40-80 mg / day, rosuvastatin 20-40 mg / day or simvastatin 40 mg / day are indicated. In absolute value, lowering LDL cholesterol by 1 mmol / l (38 mg% ml) reduces the risk of cardiovascular death and non-fatal major cardiovascular events by 20-25%11-14.
Pleiotropic effects
Acute LDL cholesterol apheresis improves endothelium-dependent vasodilation15 but other experimental studies show that the improvement of endothelial dysfunction occurs before reducing serum cholesterol level16. Pleiotropic effects of statins are not only related to the modification of blood lipids, but also to the intervention on inflammation, thrombosis, endothelial function, vascular smooth muscle cells proliferation. These effects are partly due to isoprenylated protein reduction3. These have multiple cellular functions: regulating onc genes Ras, Rac, Rho; involved in cell division, maintenance of cell shape, secretory function of the cell, cell differentiation6. Inhibition of Rho and Rho kinase isoprenylation would reduce vascular smooth muscle cell sensitivity to calcium and therefore vasospasm, would alter the cytoskeleton actin, the transmembrane and intracellular transport, the mRNA stability and should be, at least in part, responsible for the pleiotropic vascular and extravascular effects of statins15. Statins alter cell membrane rafts, the headquarters of molecules involved in immune signals. Atorvastatin, Fluvastatin, Lovastatin, Pravastatin, Simvastatin reduce the proliferation of vascular smooth muscle cells, improve endothelial vasodilation, reduce cytokine production. Platelet aggregation is reduced by atorvastatin, uninfluenced or reduced by pravastatin and simvastatin, reduced or increased by lovastatin; no data for fluvastatin3. The mechanism would consist in the reduction of the synthesis of thromboxane A2, and the amount of cholesterol in the platelet membrane16. Fibrinogen level decreases with fluvastatin, decreases or is not influenced by pravastatin and simvastatin and may increase with atorvastatin and lovastatin. PAI-1 level increases with atorvastatin, fluvastatin, simvastatin, lovastatin and decreases with pravastatin. Lipoprotein a is increased by pravastatin and simvastatin and decreased by lovastatin3. Monocyte adhesion to vascular endothelium is lowered by atorvastatin, fluvastatin, lovastatin, simvastatin. High sensitivity CRP level is reduced or unaltered by atorvastatin and simvastatin and decreases with pravastatin. All statins reduce LDL oxidation. Statins stabilize plaque by reducing its size, but especially by anti-inflammatory action: reduce macrophage accumulation and metalloproteinases activity, reduce IL-6, IL-8, IL-1, TGF-beta, NF-kB, matrix metalloproteinases17, inhibit ICAM, CD18, CD4918, block expression major histocompatibility system II (HLA type 2) under the action of IFN-gamma on endothelial cells, macrophages, microglia. They are immunosuppressants, with potential clinical implications in transplant. Experimentally, statins are proangiogenic in low doses and antiangiogenic, including in tumors, at high doses3. Statins inhib plaque angiogennesis18. They have dual effect on endothelial apoptosis and prevent senescence by altering isoprenylated proteins and by influencing the telomerase. Clinical translation of these experimental data is difficult. The variability of pleiotropic effects depends on many factors, including lipophilicity of molecules. Lipophylic statins have extrahepatic distribution and would have more important pleiotropic effects compared to the hydrophilic statins, with predominant hepatic distribution. However, there are proofs of pleiotropic effects for pravastatin and rosuvastatin whith limited extrahepatic distribution. In experimental and clinical studies14, rosuvastatin improves endothelial function, reduces adiposity and hepatic steatosis and pravastatin influences endothelial dysfunction and platelet aggregation6. The clinical relevance of the distinction between fat soluble and water soluble statins in terms of pleiotropic effects is not clear.Other actions have also been described: simvastatin ameliorates renal fibrosis19; statins are useful in preventing vasospasm after aneurysm rupture cerebral hemorrhage20; when administered preoperatively, they reduce the risk of postoperative acute renal failure21. Statins can prevent atrial fibrillation, have favorable effects in pneumonia, protect against nuclear cataract. They prevent ischemic stroke, probably less by reducing serum cholesterol, as by increasing NO dependent cerebral vasodilatation22.
Side effects
A meta-analysis that included 107 835 patients treated with statins (simvastatin and atrovastatin in 53% patients) showed that 17.4% of patients had at least one adverse effect: 27% myalgia or myopathy versus 4.7% in the general population, 2.5% other changes in muscle or connective tissue, 2.3% impaired general condition, 2.1% liver damage, 1.6% gastrointestinal disorders. In total, 0.006% patients treated with statins developed rhabdomyolysis and 0.06% memory loss23,24. Myopathy can occur between several days and two years after the onset of therapy and the transaminases usually increase in the first three months. Both are reversible upon treatment discontinuation13. 1.7% patients discontinued the treatment. Of these, 40% had resumed statins, some the same drug; adverse effects reappeared in 13% of them. The adverse effects are more frequent with high doses of statins10,25,26. At least some of the adverse effects are due to the interference with mitochondrial respiratory chain and reduced ubiquinone synthesis. It was shown that serum levels of ubiquinone are reduced and plasma lactate / pyruvate ratio is increased during statin therapy, which suggests mitochondrial dysfunction27. Side effects depend on the pharmacological particularities, drug doses, genetics, drug interactions21. According to some studies, hydrophilic statins (fluvastatin, rosuvastatin, pravastatin) are less toxic than lipophilic statins (atorvastatin, lovastatin, and simvastatin). The risk of myopathy is lower with pravastatin and fluvastatin. Statins reduce the prevalence of stroke22 but the relationship with cognitive abilities is not yet clear. Statins increase the risk of diabetes dose-dependent at people with risk factors for diabetes but they are indicated for cardiovascular protection in patients with existing diabetes mellitus12,13. By treating 255 patients with statins for four years, there is a case of diabetes, but 5.4 coronary events are prevented10. The relationship with cancer was controversial. Currently they are considered to have a protective role on colon cancer, breast cancer, lymphoma (men), if administered for over a year28.
Drug interactions
Drug interactions depend largely on CYP450 pathway metabolism (Table 2)5.
Table 2. Drug interactions according to the CYP(a) metabolism
| Increase side effects CYP3A4 inhibitors | CYP3A4 | CYP2C9 | Other metabolic pathways | |
| simvastatin atorvastatin lovastatin |
Fluvastatin | Pravastatin | Rosuvastatin CYP2C19 |
|
| erythromycin | + | – | – | – |
| clarithromycin | + | – | – | – |
| telithromycin | + | – | – | – |
| warfarin | + (-atorvastatin) | -/+ | – | -/+ |
| cyclosporine | + | + | – | -/+ |
| itraconazole | + | – | – | |
| ketoconazole | + | – | ||
| fluconazole | – | + | – | – |
| HIV antiretroviral therapy | + | + | – | + |
| danazol | + | – | – | |
| diltiazem | + | – | – | – |
| verapamil | + | – | – | – |
| amiodarone | + | – | – | – |
| fibrates | + | + | – | – |
| grapefruit juice | + | – | – | – |
| aCYP – cytochrome | ||||
The simvastatin doses should not be higher than 10 mg / day in combination with amiodarone, verapamil, diltiazem and 20 mg, in combination with amlodipine and diltiazem10. The dosage of lovastatin should not exceed 20 mg / day in combination with diltiazem, verapamil and 40 mg with amiodarone10. Atorvastatin does not interact with warfarin. The effect of digoxin is enhanced by atorvastatin and uninfluenced by rosuvastatin and pravastatin. Simvastatin and lovastatin increase slightly the effect of digoxin10. Clarithromycin increases the concentration of pravastatin, inhibiting its membrane transporters5. Cyclosporine increases blood levels of pravastatin, rosuvastatin and pitavastatin through the inhibition of membrane transporters5. Rifampin and carbamazepine are inducers of CYP 3A4 and reduce the concentration of simvastatin, lovastatin and atorvastatin. They reduce the concentration of fluvastatin and pravastatin by inducing transport proteins OATP1B1, MRP2. It is not known by which mechanism they reduce the concentration of rosuvastatin and pitavastatin5. In general this interaction has no clinical relevance5. Gemfibrozil inhibits CYP2C8 and liver transmembrane transporter OATP1B1 of acid statins. It increases blood levels of atorvastatin, rosuvastatin, pravastatin, pitavastatin. It does not influence the blood level of simvastatin, lovastatin, fluvastatin5,29. The association of gemfibrozil with fluvastatin or pravastatin would lower the risk of adverse effects than other statins. Bezafibrate and fenofibrate do not affect serum levels of statin5 but the association cumulates adverse effects. Atorvastatin reduces clopidogrel effects inhibiting its activation by the CYP3A4 pathway. This interaction does not appear to have clinical relevance10. High doses of simvastatin and atorvastatin increase the blood level of digoxin by 20%, inhibiting MDR1 carrier membrane proteins5,29.
Genetics and adverse effects of statins
Lovastatin induces atrogin-1 gene expression, which would initiate myopathy6. The variability of the levels of rosuvastatin in 165 pts is correlated to 2 genetic polymorphisms: SLCO1B1carrier gene (p <0.001); ABCC2 carrier gene. SLCO1B1 gene regulates the expression of membrane transporter OATP1B1 (organic anion transporting polypeptide) involved in hepatocyte uptake of statins, especially those hydrophilic (pravastatin, rosuvastatin)30. ABCC2 gene encodes MRP2 (multidrug resistance protein 2 associated) transporter protein located on the membrane of hepatocytes at the biliary pole, involved in biliary excretion of statins5,30. Variability of serum levels of atorvastatin in 134 pts is explained by two genetic polymorphisms of SLCO1B1 gene and by the activity of CYP3A30. Two variants of SLCO1B1 were associated with myopathy on simvastatin30. SLCO 1B1 polymorphism is associated with higher levels of simvastatin, pravastatin, rosuvastatin, but not of fluvastatin5.
Conclusions
There is no doubt about the efficiency of the statins in cardiovascular prevention but still many unresolved clinical problems remain. Could statins be exchanged between them? Apparently not, their ability to reduce LDL cholesterol level is different and appropriate medications must be used in order to obtain the proposed target. Pharmacological differences between statins may influence their effects. Is it important the administration as prodrug (lactone) or active form (acid)? During their metabolism, statins pass from the lactone to the acid form and vice versa; it is hard to say whether form of administration influences the effects. Is it clinically relevant the difference between lipophilic and hydrophilic statins? Hydrophilic statins would have less adverse effects. On the other hand, side effects are correlated with the dose and the potency of drugs. Hydrophilic statins would have lower pleiotropic effects, their distribution in peripheral tissues being limited. Are pleiotrophic effects important in clinical practice? Statins largely differ between them regarding these effects but all have proofs of clinical efficiency.Statins may be discontinued? Apparently no. A study of patients with acute coronary syndromes treated with statins and followed 23 months showed that non-adherence to statins increased the rate of cardiovascular mortality approximately 3-fold. However, for primary prevention, some studies show that statins can be administered intermittently in case of intolerance. Concomitant administration of Q coenzyme can limit the adverse effects of statins? Studies are not clear in this regard. Is genetic testing useful in the treatment with statins? Probably yes, but we need more studies. The mechanisms long-term regulating cholesterol synthesis have a role in the effectiveness of statin therapy? It would be an interesting issue, but so far there are no studies in this regard.
Acknowledgement: This paper is supported by the Sectoral Operational Programme Human Resources Development (SOP HRD) 2007-2013, financed from the European Social Fund and by the Romanian Government under the contract number POSDRU/107/1.5/S/82839
References:
1. Joyce J Diwan: Biochemistry of Metabolism. 1998-2008, http:// www.rpi. edu/ dept/ bcbp/molbiochem/MBWeb/mb1/MB1index.html
2. Akira E, Kuroda M, Tsujita Y: “ML-236A, ML-236B, and ML-236C, new inhibitors of cholesterogenesis produced by Penicillium citrinium”. Journal of Antibiotics (Tokyo). 1976;29:1346–8. doi:10.7164/ antibiotics. 29.1346. PMID 1010803.
3. Patrizzia Gazzerro, Maria Chiara Proto, Giuseppina Gangemi: Pharmacological Actions of Statins: A Critical Appraisal in the Management of Cancer. Pharmacological Revue. 2012;64:102-146.
4. Srinivasa Rao K, Prasad T, Mohanta GP, Manna: An Overview of Statins as Hypolipidemic Drugs. IJPSDR. 2011; 3: 178-83
5. Neuvonen PJ, Niemi M, Janne Backman: Drug interactions with lipid-lowering drugs: Mechanisms and clinical relevance. Clinical Pharmacology and Therapeutics. 2006; 80: 565-575
6. Pang H Chong: Therapeutic Controversies; Lack of Therapeutic Interchangeability of HMG-CoA Reductase Inhibitors. Ann Pharmacother. 2002; 36:1907-1917
7. Lennernäs H and Fager G: Clinical Pharmacokinetics and Pharmacodynamics of HMG-CoA reductase inhibitors: similarties and dissimilarties. Clin Pharmacokin. 1997; 32: 403-425
8. Katharine Howe, Faizah Sanat, A E Thumser, Tanya Coleman, N Plant:. The statin class of HMGA CoA reductase inhibitors demonstrate differential activation of the nuclear receptors PXR, CAR and FXR, as well as their downstream target genes: Xenobiotica; the Fate of Foreign Compounds in Biological Systems 2011, 41 (7): 519-29
9. The Lipid Research Clinics Coronary Primary Prevention Trial results. I. Reduction in incidence of coronary heart disease. JAMA. 1984 Jan 20;251: 351-64
10. Opie LH, Gersh BJ. Class indication for Statins in Drugs for the Heart, eight edition, Elsevier-Saunders, 2013: 411-423
11. Stone NJ, Jennifer Robinson, Alice T Lichtenstein, et al.: 2013 ACC/AHA Guideline on the treatment of Blood Cholesterol to Reduce Cardiovascular Risk in Adults: A Report of the American College of Cardiiology /American Heart Association Task Force on Practice Guidelines Circulation, , http://circ:.ahajournals.org/content/early/2013/11/01.0000437738.63853.7acitation/;
12. Gielen S and Landmesser U. The Year in Cardiology:cardiovascular disease prevention. European Heart Journal. 2014; 35: 307-312
13. Ebrahim Shah, Taylor Fiona C, Brindle P. Statins for the primarry prevention of cardiovascular disease. BMJ. 2014; 348: .g280
14. Neto-Ferrera R, Rocha VN, Souza- Mello V, Mandarini de Lacerda, de Carvalho JJ: Pleiotropic effects of rosuvastatin on the glucose metabolism and the subcutaneous and visceral adipose tissue in C57B1/6 mice. Diabet Metab Syndrome. 2013; 5:32
15. Anderson TJ, Mereddith IT, Yeung AC, Frei B, Selwyn AP, Ganz: The effect of cholesterol lowering and antioxidant therapy on endothelium dependent coronary vasomotion. N. Engl J Med. 1995; 332:488-493
16. Takemoto M, Liao J K: Pleiotropic Effects of 3-Hydroxi-3 methylglutaryl Coenzyme A Reductase Inhibitors. Atherosclerosis, Thrombosis, and Vascular Biology. 2001; 21:1712-1719
17. Weitz-Schmidt G. Lymphocyte function-associated antigen-1 blockade by statins: molecular basis and biological relevance. Endothelium. 2003; 10: 43-7.
18. Yoshifumi Hamasaki, Kent Doi, Koji Okamoto, Hideaki Ijichi, George Seki, Rui Maeda-Mamiya, Toshiro Fujita, Eisei Noiri: 3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitor simvastatin ameliorates renal fibrosis through HOXA13–USAG-1 pathway. Laboratory Investigation. 2012;92, 1161-1170
19. Jason L, Trimble and Denise R Kockler. Statin Treatment of Cerebral Vasospasm after Aneurysmal Subarachnoid Hemorrhage. Ann Pharmacother. 2007; 41: 2019-2023
20. Steven M. Brunelli, Sushrut S. Waikar, Brian T. Bateman, Tara I. Chang, Joyce Lii, Amit X. Garg, Wolfgang C. Winkelmayer, Niteesh K. Choudhry.: Preoperative Statin Use and postoperatory Acute Kidney Injury. The American Journal of Medicine. 2012; 125: 1195-1204
21. Huabing Zhang, Jorge Plutzky, Stephen Skentzos, Fritha Morrison, Perry Mar, Maria Shubina, Alexander Turchin: Discontinuation of Statins in Routine Care Settings: A Cohort Study. Ann Intern Med. 2013; 158: 526-534.
22. Nickole N Henyan, Daniel M Riche, Honey E East, Pamela N Gann: Impact of Statins on Risk of Stroke: A Meta-Analysis. Ann Pharmacother. 2007 41: 1937-1945
23. EJ Mills, P. Wu, I. Chong G, Ghement I, Singh S, Akl EA, Eyawo O, Guyatt G, Berwanger O, Briel M: Efficacy and safety of statin treatment for cardiovascular disease: a network meta-analysis of 170255 patients from 76 randomized trials. QJM. 2011; 104 : 109-24.
24. Wouter Jukema, Christopher P. Cannon,Anton J. M. de Craen, Rudi G. J. Westendorp, Stella Trompet: The Controversies of Statin Therapy-Weighing the Evidence. JACC. 2012; 60: 875-881
25. Jaakko Allonen, Markku S. Nieminen, Maisa Lokki,Olavi Parkkonen, Satu Vaara, Markus Perola, PhD; Tero Hiekkalinna, Timo E. Strandberg, Juha Sinisalo: Mortality Rate Increases Steeply With Nonadherence to Statin Therapy in Patients With Acute Coronary Syndrome. Clin. Cardiol. 2012; 35: E22–E27
26. Erin K Reindl, Wright BM, and Wargo KA: Alternate-Day Statin Therapy for the Treatment of Hyperlipidemia. The Annals of Pharmacotherapy. 2010,44:1459-1470
27. de Pinieux, P. Chariot, M. Ammi-Sai, D.F. Louarn , J. L. Lejonc A , Astier B. Jacotot, R. Gherardi: Lipid-lowering drugs and mitochondrial function: effects of HMG-CoA reductase inhibitors on serum ubiquinone and blood lactate/pyruvate ratio. Br J Clin Pharmacol. 1996; 42: 333–337
28. Lutski M, Shalev V, Porath A, Chodick G: Continuation of statin therapy and the risk of primary cancer : a populatio-based study. Prev Chronic Dis. 2012; 9:E137.
29. Bellosta S, Paoletti R, Corsini A: Safety of Statins. Focus on Clinical Pharmacokinetics and Drug Interactions. Circulation. 2004; 109 (suppl lll): lll-50-lll -57
30. Stiles S: Pharmacogenetics and Statins: Genotyping Might Cut Muscle-Pain Risk 2013; www.medscape.com/viewarticle/809552
 This work is licensed under a
This work is licensed under a