Christian Wolpert1, Maja Vogel1, Christian Nagel1, Norman Rüb1
1 Department of Medicine – Cardiology, Klinikum Ludwigsburg, Germany
Keywords: Short QT, Sudden Death, atrial fibrillation, congenital, arrhythmia
INTRODUCTION
In 2000 Gussak et al. first described an idiopathic short QT interval associated with atrial fi brillation (AF) in one family and a sudden death in an unrelated individual1. Three years later, in 2003, Gaita et al. reported the association of a short QT interval and sudden cardiac death in two unrelated European families1. In recent years a variety of mutations in different genes most likely causative for the short QT interval were identifi ed. The initially reported mutations either caused a gain-of-function of cardiac potassium channels IKr, IKs and IK1, or a loss-of-function in the cardiac L-type calcium channel (ICa)3-10. Meanwhile new mutations have been reported resulting in different alterations of ion channel activity. Risk of sudden death in patients with a Short QT syndrome has been reported to be high. Also the occurrence of atrial fi brillation at younger ages is not infrequently seen in these patients, most likely caused by short atrial refractory periods.
CLINICAL PRESENTATION
The clinical presentation of patients with SQTS is very heterogeneous. First data were presented from the EUROSHORT registry10. 29 patients (21 m, 8 f) were studied. 18/29 were symptomatic at time of enrolment. Nine of the patients had a history of cardiac arrest, 6 had suffered syncope and 7 had documented atrial fl utter or atrial fi brillation10. Onset of symptoms was very variable ranging from the age of 4 months up to the age of 62 years and it was distributed over all decades of life. Sudden deaths occurred in the youngest patient at the age of 4 months. thus, SQTS represented also a new cause for the sudden infant death syndrome (SIDS). Mazzanti et al. studied a population of 47 probands who were referred to the database for cardiac arrest (n=19), syncope (n=9), family history of sudden death (n=2) or an incidentally found short QTc interval11. 12/47 had a family history of sudden death in the young and 4 had multiple victims in their family (2.5±0.6.) Among the asymptomatic individuals, the sudden death victims and patients with syncope the QTc interval were not statistically signifi cantly different. The age at time of syncope or sudden death was also comparable with 21±11 vs 25±13 years. Interestingly, the QTc interval in those, in whom a mutation was identifi ed, was signifi cantly shorter (300 vs 335 ms). There was no difference in the likelihood of sudden death between mutation positive and mutation negative probands. Villafane et al. published a multicentric series of 21 pediatric patients12,13. The median age was 15 years. (84% males) 56% of the patients were symptomatic for syncope (n=4) or sudden death (n=6). 16 patients had either a personal or family history of sudden death. The rate of atrial fi brillation was very high for this young cohort with 4/21 patients. A gene mutation was identifi ed in only 24%. Eleven of 21 patients received an ICD and two patients received an appropriate shock and 64% inappropriate shocks. The authors applied the Gollob score and observed that asymptomatic individuals with a Gollob score of <5 were asymptomatic for VT/VF or sudden death and syncope over a 6 year follow-up. In a Japanese series of fi ve Japanese unrelated families symptoms were AF in 2, ventricular fi brillation (VF) in 2, sudden death in 3 patients and severe bradycardia in one new-born. The QTc in this series was between 280 and 340 ms i.e. somewhat longer than in patients with SQT114,15.
ELECTROCARDIOGRAPHIC FINDINGS
There is no clear definition of a short QT interval, but a number of authors and the ESC guideline committee has come up with a proposal on where the lower limit of the QT interval should be.
In the following there are some of the works that have tried to determine the upper and lower boundaries of the QTc interval. In the general population QTc intervals follow a Gaussian normal distribution16-23. Normal QT intervals were proposed as QTc intervals within two standard deviations from the mean. QTc shorter than the 2.5th percentile were defi ned as “short”. Following this calculation, QTc of <350 ms for men and QTc <360 ms for women are considered short. In large population based studies the prevalence of a short QT interval was analysed (Anttonen et al.). 10.822 subjects and found short QTc intervals of <340 ms in 0.4% of the subjects16. Extremely short QTc intervals <320 ms were seen in 0.1% of the cases. Both, individuals with a short and a very short QTc interval had no cardiac events8. In a Japanese cohort of 12.149 subjects 0.01% exhibited a QTc interval within the 2.5th percentile (men QTc <354 ms; females <364 ms) and only 3 male subjects a QTc of <300 ms17. Another analysis of 19.53 subjects undergoing biannual health examinations in the follow-up program in Hiroshima and Nagasaki since 1958 the prevalence for a short QT interval (QTc <350 ms) was 0.01%13. Kobza et al. found a similar low prevalence of 0.01% of QTc intervals <320 ms in 41.767 male army conscripts19. A recent report on an ECG population sample among 1.7 million persons revealed a QTc of less than 300 ms in 2.7 in 100.000. The risk of dying over a follow up of 8.3 years of follow-up was increased 2.6 fold21.
The electrocardiogram of the fi rst patients identifi ed with a SQTS (SQT1) showed very short QT intervals and in addition short QT intervals corrected for heart rate (QTc <300 ms). The patients identified as SQT2 – SQT5 exhibited QTc of up to 360 ms. The ECG in SQT1-3 reveals tall, symmetrical and asymmetrical peaked T wave especially in the precordial leads. In most cases a ST segment is absent with the T wave originating directly from the S wave. Another fi nding in SQTS is a prolonged Tpeak – Tend interval. Anttonen et al. compared the Jpoint-Tpeak interval in symptomatic patients with SQTS, probands with a short QT interval and a control group of subjects with normal QT interval24. Symptomatic patients with SQTS had significantly shorter Jpoint-Tpeak intervals and higher corrected Tpeak-Tend/QTc ratio compared to asymptomatic probands with a short QT interval and subjects with a normal QT interval. Patients diagnosed with SQT4 and SQT5 and carry a mutation in the cardiac calcium channel exhibit shorter than normal QT intervals of 330-360 ms, which is relatively longer than in SQT1-SQT3. These patients additionally displayed J point elevations diagnostic of Brugada syndrome5. Villafane et al. reported the data on pediatric patients with a short QT syndrome. The QTc ranged here from 194 to 355ms (mean 312 ms)12,13.
QT adaptation to heart rate and behaviour during exercise
Another important finding in the initially reported SQT1 patients was the inappropriate adaptation of the QT interval to heart rate. In the fi rst patients with the KCNH2 mutation Wolpert et al. could show that the QT interval did not shorten appropriately compared to normal controls. Treatment with quinidine was able to restore the QTc/heart rate ratio towards the normal range10,25. Giustetto and co-workers further studied the usefulness of exercise testing in the diagnosis of short QT in order to see if QT behaviour during exercise helps to differentiate between short QT patients and individuals with a shorter than normal QT interval. They investigated twenty one patients with a short QT syndrome and matched controls. Rest and peak exercise heart rates did not differ between the groups. The baseline QT intervals at rest were 276 vs 364 ms and at peak exercise 228±27 ms vs. 245±26 ms with a mean variation from rest to peak exercise of 48±14 vs. 120±20 ms. The QT/HR slope never exceeded 0.9 ms/ bpm. The mean was -0.53 ms/bpm vs. -1.29 ms/bpm.
Electrophysiological findings
Atrial and ventricular effective refractory periods are signifi cantly shortened especially in SQT1 (KCNH2)29. Atrial refractory period of 140 ms and ventricular effective refractory period of 150 ms or less are criteria highly suspicious of the SQTS. Inducibility of ventricular fi brillation during programmed ventricular stimulation is high in patients with SQTS 116. The current consensus recommendations and the ESC guidelines however do not recommend programmed ventricular stimulation for diagnostic purposes or risk stratifi cation. It is only done within research protocols.
RISK STRATIFICATION
The QTc intervals of these patients are ranging from <300 ms up to <360 ms. In summary, a short QT interval on the 12-lead ECG does not predict a risk for life threatening tachy-arrhythmias per se. However, the rare fi nding of a short QT interval should initiate a diagnostic work-up including family members. In the case of a short QT interval together with episodes of atrial fibrillation, sustained palpitation, unexplained syncope, ventricular fi brillation, and/or a positive family history for premature sudden cardiac death, Short QT syndrome should be suspected30,31. Gollob et al proposed a SQTS Diagnostic criteria score analogous to the Schwartz-Score for Long QT syndrome in which a high probability of SQTS was reached when more or equal to 4 points were given. In his criteria a QTc of <370 ms was 1 point, <350 ms 2 points and <330 ms equal to 3 points31. The 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death provide a class I recommendation for a diagnosis of a SQTS, when the QTc is <340 ms. It should be considered, if the QTc is <360ms and one or more of the following conditions exists a) a confi rmed pathogenic mutation, b) a family history of Short QT syndrome, c) a family history of sudden death at age <40 years or d) survival from a VT/VF episode in the absence of heart disease. The guidelines discourage with a class III indication the EP study for risk stratification30.
MOLECULAR AND GENETIC MECHANISMS
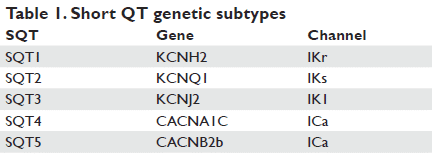
The SQTS is a genetically heterogeneous disease like long QT syndrome. A number of genes have been described to be associated with Short QT syndrome. The mutations are located on different chromosomes e.g. 7, 10, 11, 12 and 17 and encode for different cardiac ion channels. The first mutation identifi ed to be causing short QT syndrome (SQT1) was a gain of function mutation leading to an increase of the rapid component of the delayed rectifi er potassium current (IKr)7. Two different missense mutations were identifi ed resulting in the same amino acid change in HERG (KCNH2). These mutations at nucleotide 1764 in the KCNH2 gene substitute the asparagine at codon 588 for a positively charged lysine (p.N588K). The p.N588K mutation causes a loss of the normal rectifi cation of the current at plateau voltages, which results in a signifi cant increase of IKr during phase 2 and 3 of the action potential leading to abbreviation of the action potential and both, atrial and ventricular refractoriness. Bellocq et al. shortly after reported on a mutation in a single sporadic case of a 70-year-old patient with SQTS (QTc 302 ms) and sudden cardiac arrest. They identifi ed a gain of function mutation (p.V307L) in the KCNQ1 gene which encodes the slow component of the delayed rectifi er potassium channel (IKs) (SQT2). A further missense mutation in the same was identified in a baby with bradycardia and atrial fi brillation in utero6. The ECG of the new-born revealed a shortened QT interval and episodes of atrial fibrillation. Priori and co-workers later identified in two relatives without sudden cardiac arrest a gain of function in KCNJ2, encoding the inward rectifier potassium channel (IK1) causing abbreviation of the QT interval and asymmetrical T waves with a rapid terminal downslope4. Later, we, together with Antzelevitch and co-workers, further described novel mutations of the cardiac L-type calcium channel genes responsible for shortening of the QT interval in families characterized by sudden cardiac death, atrial fi brillation together with a Brugada type I ECG pattern5. Functional analyses revealed loss-of-function missense mutations of the CACNA1C and CACNB2b genes encoding the pore forming of Cav1.2 1- and 2b-subunits of the cardiac L-type calcium channel. The decreased net current of the cardiac L-type calcium channels led to an abbreviation of the plateau phase of the action. Recently new patients have been identifi ed who suffer from mutations in KCNH2 and KCNQ1. A Japanese group and a Chinese group identifi ed a novel mutation in KCNH2 that resulted in a 2.5-fold increase in peak current density in COS-7 cells and a mutation in KCNH2-p.T618L14. Moreno et al presented a male individual with a family history of sudden death with a QTc of 356 ms who carried a mutation in the S5 segment of the KCNQ1 that impaired its association with KCNE19. Suzuki reported a case of a asymptomatic 10-year old boy who displayed a QTc interval of 260 ms. In molecular genetic screening they found a mutation in the KCNH2-p.N588K, identical to the one identifi ed in the fi rst two unrelated families in 2000 and 2003 when the syndrome was described fi rst in detail32. Deo et al described a mutation in KCNJ2 that resulted in an enhanced IK1 outward current leading to a phenotype of an extremely abbreviated QT interval and atrial fi brillation33. A French group presented a family with inherited Lcarnitine defi ciency, in which a short QT interval was found in all affected members. After substitution of carnitine the QT interval was signifi cantly prolonged towards normal range34.
PHARMACOLOGIC THERAPY OF SHORT QT SYNDROME
Most of the experiences in vitro and in vivo are available for patients with SQT1. Heterogeneous expression studies exhibited that the p.N588K mutation increased the density of IKr and reduced the affinity of IKr blockers like d-sotalol 20-fold. McPate et al. could demonstrate that the effect of E-4031, a specific IKr blocker, was also signifi cantly attenuated by the p.N588K mutation, whereas quinidine was less and disopyramide the least affected by p.N588K-HERG40. Cordeiro et al. could nicely show that these fi ndings are based on the +90 mV shift in the voltage-dependence of inactivation of the HERG channels. Most IKr-blockers interact with the HERG channels in the inactivated state. Thus, a failure of inactivation of the HERG channel leads to the ineffi cacy of the specifi c IKr blockers41. Recently, McPate et al. could demonstrate that besides disopyramide and quinidine also propafenone and amiodarone were only slightly inhibited by the mutant p.N588K41. Thus these drugs may represent an additional option in the pharmacologic treatment of SQT1. In vivo, several class I and III antiarrhythmic drugs have been tested in patients with a mutation in HERG (SQT1). Neither d-sotalol nor ibutilide were able to prolong QT interval in the fi rst series of type I Short QT syndrome patients. Flecainide, a Na+-channel blocker, which has in addition a blocking effect on IKr and on the transient outward potassium current (Ito), led to a slight increase in ventricular effective refractory periods, but failed to effectively prolong the QT interval42. In contrast quinidine was able to normalise the QT interval and to prolong the ventricular effective refractory period in patients with a SQT129. Further quinidine restored the heart rate dependence of the QT interval towards the normal range and rendered ventricular tachyarrhythmias non-inducible in patients in whom baseline electrophysiological studies demonstrated reproducible inducibility of ventricular fi brillation. Following the positive effects of disopyramide in in vitro experiments disopyramide has also been shown to be effective in a pilot study in patients with a SQT1 by Schimpf et al. The most frequently used drug for treatment of VF prevention or recurrences was quinidine. No patient on quinidine therapy suffered from ventricular fibrillation or a recurrence of atrial fi brillation during midterm follow-up in the Short QT registry10,38. A subset from one SQT-1 family published by Bjerregaard et al. treated with propafenone is free of recurrences of atrial fi brillation without prolongation of the QT interval. However, it is not known if the fact, that they did not suffer from ventricular fi brillation during follow up is caused by the effect of the drug or incidental (personal communication). Whether the effects of the investigated class I and III drugs can be translated to SQT2 – SQT5 is not clear. However, in a patient with SQT4 quinidine was equally able to prolong QT interval and suppress paroxysms of atrial fi brillation as seen in SQT1. Due to the electrophysiologic and genetic heterogenity of the SQTS therapy may have very different effects depending on the type of mutation and the affected channel. Further studies of pharmacologic therapy are needed to elucidate the potential long term benefit of pharmacologic treatment for both prevention of atrial fi brillation and sudden death. The 2015 ESC guidelines recommend quinidine or sotalol when patients refuse an ICD or have a contraindication and in asymptomatic patients and a family history with a class IIb indication. Finally, however, some drug combinations have been successfully used in patients, that are not mentioned in the guidelines and to some extent the treatment of this very heterogeneous patients will remain individual30.
ICD THERAPY
To date the only reliable treatment to prevent patients from sudden cardiac death is the implantation of an implantable cardioverter-defi brillator (ICD). In symptomatic patients with SQTS the ICD is the therapy of choice, while antiarrhythmic drug therapy may rethe QT interval and to prolong the ventricular effective refractory period in patients with a SQT129. Further quinidine restored the heart rate dependence of the QT interval towards the normal range and rendered ventricular tachyarrhythmias non-inducible in patients in whom baseline electrophysiological studies demonstrated reproducible inducibility of ventricular fi brillation. Following the positive effects of disopyramide in in vitro experiments disopyramide has also been shown to be effective in a pilot study in patients with a SQT1 by Schimpf et al. The most frequently used drug for treatment of VF prevention or recurrences was quinidine. No patient on quinidine therapy suffered from ventricular fibrillation or a recurrence of atrial fi brillation during midterm follow-up in the Short QT registry10,38. A subset from one SQT-1 family published by Bjerregaard et al. treated with propafenone is free of recurrences of atrial fi brillation without prolongation of the QT interval. However, it is not known if the fact, that they did not suffer from ventricular fibrillation during follow up is caused by the effect of the drug or incidental (personal communication). Whether the effects of the investigated class I and III drugs can be translated to SQT2 – SQT5 is not clear. However, in a patient with SQT4 quinidine was equally able to prolong QT interval and suppress paroxysms of atrial fi brillation as seen in SQT1. Due to the electrophysiologic and genetic heterogenity of the SQTS therapy may have very different effects depending on the type of mutation and the affected channel. Further studies of pharmacologic therapy are needed to elucidate the potential long term benefi t of pharmacologic treatment for both prevention of atrial fi brillation and sudden death. The 2015 ESC guidelines recommend quinidine or sotalol when patients refuse an ICD or have a contraindication and in asymptomatic patients and a family history with a class IIb indication. Finally, however, some drug combinations have been successfully used in patients, that are not mentioned in the guidelines and to some extent the treatment of this very heterogeneous patients will remain individual30.
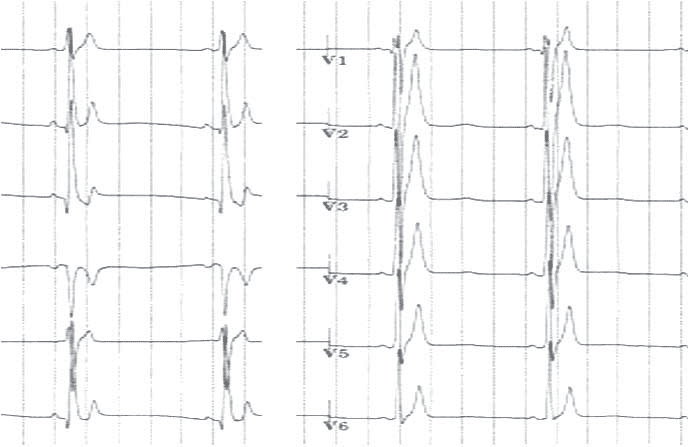
Figure 1. The figure displays the ECG of a young patient with Short QT syndrome.
SUMMARY
The SQTS is one of the primary electrical diseases of the heart with a high incidence of syncope and sudden cardiac death. The hallmark for the diagnosis is a short QT interval. Since the disease presentation is quite heterogeneous, a strong genotype-phenotype correlation and a conclusive risk stratifi cation is still very diffi cult to achieve. Patients with SQTS should be referred for genetic counselling, molecular genetic analysis and initiation of family screening in specialized centers in order not to miss affected individuals at risk or to over diagnose patients with a shorter than normal QTc who are not at risk of sudden death.
Conflict of interest: none declared.
References
1. Gussak I, Brugada P, Brugada J, Wright RS, Kopecky SL, Chaitman BR et al. (2000) Idiopathic short QT interval: a new clinical syndrome? Cardiology 94:99-102.
2. Gaita F, Giustetto C, Bianchi F, Wolpert C, Schimpf R, Riccardi R et al. (2003) Short QT Syndrome: a familial cause of sudden death. Circulation 108:965-970
3. Bellocq C, van Ginneken AC, Bezzina CR, Alders M, Escande D, Mannens MM et al. (2004) Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 109:2394-2397.
4. Priori SG, Pandit SV, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, et al. (2005) A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res 96:800-807.
5. Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, et al. (2007) Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 115:442-449.
6. Hong K, Piper DR, Diaz-Valdecantos A, Brugada J, Oliva A, Burashnikov E et al. (2005) De novo KCNQ1 mutation responsible for atrial fi brillation and short QT syndrome in utero. Cardiovasc Res 68:433-440.
7. Brugada R, Hong K, Dumaine R, Cordeiro J, Gaita F, Borggrefe M et al. (2004) Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation 109:30-35.
8. El Harchi A, Melgari D, Zhang YH, Zhang H, Hancox C. Action potential clamp and pharmacology of the variant 1 Short QT Syndrome T618I hERG K+ Channel. PLoS ONE 2012,7: e52451
9. Moreno C, Olivera A, de la Cruz A, Bartolucci C, Mun C, Salar E et al. A new KCNQ1 mutation at the S5 segment that impairs its association with KCNE1 is responsible for short QT syndrome. Cardiovascular Research 2015:107:613-623
10. Giustetto C, Di Monte F, Wolpert C, Borggrefe M, Schimpf R, Sbragia P et al. (2006) Short QT syndrome: clinical fi ndings and diagnostictherapeutic implications. Eur Heart J 27:2440-2447.
11. Mazzanti A, Kanthan A, Monteforte N, Memmi M, Bloise R, Novelli V et al. Novel Insight Into the Natural History of Short QT Syndrome. J Am Coll Cardiol 2014;63:1300–8
12. Villafane J, Atalla J, Gollob M, Maury P, Wolpert C, Gebauer R et al. Long-Term follow-up of a pediatric cohort with short QT syndrome. J Am Coll Cardiol 2013;61:1183–91
13. Villafane J, Fischbach P, Gebauer R. Short QT syndrome manifesting with neonatal atrial fi brillation and bradycardia. Cardiology 2014; 128:236–240
14. Harrell DT, Ashihara T, Ishikawa T, Tominaga I, Mazzanti A, Takahashi K et al.Genotype-dependent differences in age of manifestation and arrhythmia complications in short QT syndrome. International Journal of Cardiology 2015;190:393-402
15. Fukuyama M, Ohno S, Wang W, Kimura H, Makivama T, It oh H, et al. L-Type calcium channel mutations in Japanese Patients. With Inherited Arrhythmias. Circ J 2013;77:1799-1806
16. Anttonen O, Junttila MJ, Rissanen H, Reunanen A, Viitasalo M, Huikuri HV (2007) Prevalence and prognostic signifi cance of short QT interval in a middle-aged Finnish population. Circulation 116:714-720.
17. Funada A, Hayashi K, Ino H, Fujino N, Uchiyama K, Sakata K et al. (2008) Assessment of QT intervals and prevalence of short QT syndrome in Japan. Clin Cardiol 31:270-274.
18. Gallagher MM, Magliano G, Yap YG, Padula M, Morgia V, Postorino C et al. (2006) Distribution and prognostic signifi cance of QT intervals in the lowest half centile in 12,012 apparently healthy persons. Am J Cardiol 98:933-935.
19. Kobza R, Roos M, Niggli B, Abacherli R, Lupi GA, Frey F et al. (2009) Prevalence of long and short QT in a young population of 41,767 predominantly male Swiss conscripts. Heart Rhythm 6:652-657.
20. Viskin S (2009) The QT interval: too long, too short or just right. Heart Rhythm 6:711-715.
21. Iribarren C, Round AD, Peng J, Lu M, Klatzsky AL, Zaroff JG et al. Short QT in a Cohort of 1.7 Million Persons: Prevalence, correlates, and prognosis. Ann Noninvasive Electrocardiol 2014;19:490–500
22. Portugal G, Olivera MM, Cunha P, Ferreira F, Lousinha A, Fiarresga A et al. Short QT syndrome presenting as syncope: How short is too short? Rev Port Cardiol. 2014;33:649.e1—649.e6
23. Postema PG, Wilde AMM, The measurement of the QT interval. Current Cardiology Reviews, 2014, 10, 287-294
24. Anttonen O, Junttila MJ, Maury P, Schimpf R, Wolpert C, Borggrefe M et al. (2009) Differences in twelve-lead electrocardiogram between symptomatic and asymptomatic subjects with short
QT interval. Heart Rhythm 6:267-271
25. Giustetto C, Scorocco C, Schimpf R, Maury P, Mazzanti A, Levetto M et al. Usefulness of exercise test in the diagnosis of shortQT syndrome. Europace 2015;17:628-634
26. Tülümen E, Giustetto C, Wolpert C, Maury P, Anttonen O, Probst V et al. PQ segment depression in patients with short QT syndrome: A novel marker for diagnosing short QT Syndrome? Heart Rhythm 2014;11:1024–1030
27. Schimpf Echo
28. Frea S, Giustetto C, Capriolo M, Scrocco C, Fornengo C, Benedetto S et al. New echocardiographic insights in short QT syndrome: More than a channelopathy? Heart Rhythm 2015;12:2096–2105
29. Wolpert C, Schimpf R, Giustetto C, Antzelevitch C, Cordeiro J, Dumaine R et al. (2005) Further insights into the effect of quinidine in short QT syndrome caused by a mutation in HERG. J Cardiovasc Electrophysiol 16:54-58.
30. Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J et al. (2015) 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J 36:2793-2867
31. Gollob MH, Redpath CJ, Roberts JD (2011) The Short QT Syndrome. JACC 57:802-812
32. Suzuki H, Hoshina S, Ozawa J, Sato A, Minamino T, Aiza was Y. Short QT syndrome in a boy diagnosed on screening for heart disease. Pediatr Int 2014: 775-776
33. Deo M, Ruan Y, Pandit SV, Shah K, Berenfeld O, Blaufox A, Cerrone M et al. (2013) KCNJ2 mutation in short QT syndrome 3 results in atrial fibrillation and ventricular proarrhythmia. Proc Natl Acad Sci USA 110:4291-4296
34. Roussel J, Labarthe F, Thireau J, Ferro F, Farah C, Roy J et al. Carnitine defi ciency induces a short QTsyndrome. Heart Rhythm 2016;13: 165–174
35. Extramiana F, Antzelevitch C (2004) Amplifi ed transmural dispersion of repolarization as the basis for arrhythmogenesis in a canine ventricular- wedge model of short-QT syndrome. Circulation 110:3661- 3666.
36. Milberg P, Tegelkamp R, Osada N, Schimpf R, Wolpert C, Breithardt G et al. (2007) Reduction of dispersion of repolarization and prolongation of postrepolarization refractoriness explain the antiarrhythmic effects of quinidine in a model of short QT syndrome. J Cardiovasc Electrophysiol 18:658-664
37. Schimpf R, Bauersfeld U, Gaita F, Wolpert C (2005) Short QT syndrome: successful prevention of sudden cardiac death in an adolescent by implantable cardioverter-defi brillator treatment for primary prophylaxis. Heart Rhythm 2:416-417
38. Giustetto C, Schimpf R, Mazzanti A, Scorocco S, Maury P, Anttonen O et al. (2011) Long-term follow-up of patients with short QT syndrome. JACC 58;587-595
39. Meijer van Putten R, Mengarelli I, Guan K, Zegers JG, van Ginneken ACG, Verkerk AO. Ion channelopathies in human induced pluripotent stem cell derived cardiomyocytes: a dynamic clamp study with virtual IK1. Frontiers Physiol 2015;6:1-18
40. McPate MJ, Zhang H, Adeniran I, Cordeiro JM, Witchel HJ, Hancox JC (2009) Comparative effects of the short QT N588K mutation at 37 degrees C on hERG K+ channel current during ventricular, Purkinje fi bre and atrial action potentials: an action potential clamp study. J Physiol Pharmacol 60:23-41
41. Cordeiro JM, Brugada R, Wu YS, Hong K, Dumaine R (2005) Modulation of I(Kr) inactivation by mutation N588K in KCNH2: a link to arrhythmogenesis in short QT syndrome. Cardiovasc Res 67:498-
509
42. Gaita F, Giustetto C, Bianchi F, Schimpf R, Haissaguerre M, Calo L et al. (2004) Short QT syndrome: pharmacological treatment. J Am Coll Cardiol 43:1494-1499.
43. Schimpf R, Wolpert C, Bianchi F, Giustetto C, Gaita F, Bauersfeld U et al. (2003) Congenital short QT syndrome and implantable cardioverter defi brillator treatment: inherent risk for inappropriate shock delivery. J Cardiovasc Electrophysiol 14:1273-1277.
 This work is licensed under a
This work is licensed under a