Monica Chivulescu1, Øyvind H. Lie1, Kristina H. Haugaa1, Ruxandra Jurcut2,3
1 Center for Cardiological Innovation, Oslo University Hospital
2 „Prof. Dr. C. C. Iliescu” Emergency Institute for Cardiovascular Disease, Bucharest, Romania
3 „Carol Davila” University of Medicine and Pharmacy, Bucharest, Romania
Abstract: Arrhythmogenic cardiomyopathy is an autosomal dominant inherited cardiomyopathy that predispose to malignant arrhythmic events and heart failure. Arrhythmic risk stratifi cation is manly based on arrhythmic history and phenotype severity. Implantation of a cardiac defi brillator is the only effective strategy to prevent sudden cardiac death. Survival have been improved once implantable cardioverter-defi brillator has been extensively used and the number of ma-lignant ventricular arrhythmias survivors developing heart failure has substantially increased. Heart failure develops more often in the setting of left ventricle or biventricular involvement. Left ventricle dysfunction and heart failure are associated with specifi c genotype. Serial longitudinally follow-up of arrhythmogenic cardiomyopathy patients reported development of new criteria over time and progressive ventricular dilatation and dysfunction as well as development of full arrhythmo-genic cardiomyopathy phenotype over time in mutation- positive family members. Recognition of risk markers is important in the setting of arrhythmogenic cardiomyopathy for an optimal management.
Keywords: arrhythmogenic cardiomyopathy, arrhythmias, heart failure, sudden cardiac death, risk markers
INTRODUCTION
Arrhythmogenic cardiomyopathy (AC), an autoso-mal dominant inherited cardiomyopathy, results from desmosomal gene mutations, mechanical uncoupling, cell detachment, progressive myocytes death and sub-sequent fi brofatty tissue replacement of the ventricu-lar myocardium1.
Natural history of the disease has been derived from outcome studies which showed that AC has 2 main outcomes: malignant arrhythmic events (sud-den cardiac death (SCD), ventricular fi brillation(VF)/ sustained ventricular tachycardia (SVT), appropriate implantable cardioverter-defibrillator (ICD) therapy) and more rarely heart failure (HF) (end-stage HF, HF death and cardiac transplantation). Several clinical phases of the disease have been described: a concealed phase with subtle structural changes and risk of sudden cardiac death, a second phase of clinical overt disease with right ventricle (RV) functional and structural abnormalities in addition to arrhythmias, a third phase of severe RV dilatation and dysfunction with right HF and preserved left ventri-cle (LV) function and a forth phase of biventricular involvement and dysfunction which mimic dilated car-diomyopathy2. In addition, there have been described 3 pattern of disease expression: classic pattern with isolated RV disease and LV involvement in the advanced phase of the disease, predominant LV involvement form and biventricular pattern with paralleled biventricular im-pairment3.
AC has a heterogeneous progression, from long-term favorable course to ventricular life-threatening arrhythmias and progressive right and biventricular dilatation and dysfunction fi nally leading to HF.
EVALUATION OF ARRHYTHMIC RISK IN ARRHYTHMOGENIC CARDIOMYOPATHY
As electrical instability is present from early disease phases, cardiac arrest due to malignant arrhythmias may occur as the fi rst manifestation of the disease in young, previously healthy and asymptomatic individu-als. Studies that sought predictors of life threatening arrhythmic events had different designs4. Many stu-dies sought predictors of arrhythmic outcome as a composite measure of SCD, resuscitated SCD, VF/ SVT, appropriate ICD therapy5-7. Other studies focu-sed on identifying predictors of appropriate ICD the-rapy in patients with primary prevention implanted ICD8,9 or in patients with both primary and secondary implanted ICD10-12. There were also research groups that studied the composite outcome of cardiovascular death13 or the composite outcome of major adver-se cardiovascular events such as cardiac arrest, heart transplantation, survived SCD, VF, SVT and arrhyth-mic syncope14-16.
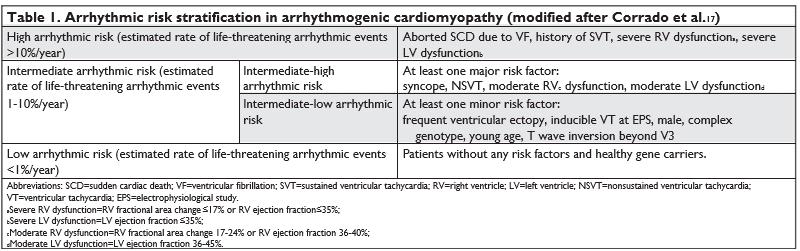
Arrhythmic risk stratifi cation is manly based on arrhythmic history and phenotype severity. An in-ternational task force consensus document defi ned 3 groups of patients according to their arrhythmic risk 17 (Table 1). High-risk patients received a class I indication of ICD implantation, intermediate-high group received a class IIa indication of ICD implan-tation, intermediate-low group received a class IIb indication and low-risk patients received a class III indication of ICD implantation17. A recent study18 eva-luated the performance of the International Task For-ce Consensus Statement Risk Stratifi cation Algorithm17. This study concluded that the algorithm accurately differentiated survival from any sustained ventricular arrhythmia (VA) among different categories although the incidence rate was higher than estimated for pa-tients in class I and class IIa indication; the algorithm did not differentiated survival free from ventricular fi brillation/fl utter between high risk and intermedia-te-high risk groups18. Nevertheless, more important than differentiation of arrhythmic risk between class I and class IIa is to defi ne patients who have a low enough arrhythmic risk so that ICD implantation can be deferred and unnecessary complications avoided19.
While there is general agreement that a history of resuscitated sudden cardiac death and VF/hemodyna-mically unstable VT confers the highest risk of SCD, the association of other risk markers with SCD de-pend on the studied population20. Regardless of the phenotypic expression, male sex, syncope, T-wave inversions beyond V3, RV dysfunction and NSVT/SVT are risk markers of arrhythmic outcome. History of exposure to strenuous exercise and VT inducibility at electrophysiological study are additional risk markers in borderline AC patients. In AC mutation carriers, the list of arrhythmic predictors is extended by inclu-ding the presence of symptoms, multiple mutations, ventricular ectopy and LV dysfunction20.
Conventional echocardiographic methods are use-ful in assessing regional wall motion abnormalities, global RV dysfunction and RV dilatation and global LV dysfunction and LV dilatation21. Alternatively, the same parameters can be assessed by cardiac magnetic resonance (CMR) with higher spatial resolution22. In addi-tion, ventricular function can be assessed by speckle tracking echocardiography21. Peak systolic myocardial strain by 2D speckle tracking echocardiography from 3 RV free wall segments is averaged as a measure of RV longitudinal strain (RVLS) and from 16 LV segments is averaged as a measure of LV global longitudinal strain (LV GLS). Alternatively, RV peak systolic longitudinal strain values from 6 segments are averaged to calcu-late RV global longitudinal strain (RV GLS). This has lower absolute values compared with RVLS21. Tem-poral parameters such as time-to-peak strain can be also assessed. Mechanical dispersion is measured as the standard deviation of the time from onset R on ECG to maximum LV and RV shortening by strain in a 6 RV segment and a 16 LV segment model respec-tively. Diagnosis of the disease and risk stratification are a composite of familiar, ECG, arrhythmic history, histological, functional and structural features21.
Thus, future guidelines regarding management of AC patients need to include a calculator of arrhythmic risk based on individual characteristics of the patient that can help the clinician to make decisions regarding ICD implantation.
Implantation of a cardiac defi brillator is the only effective strategy to prevent SCD while antiarrhyth-mic and ablation therapy improve symptoms and qua-lity of life by reducing arrhythmic recurrences and ICD discharges23. Thus, reducing the risk of SCD by properly risk-stratifying patients who would benefi t from an ICD is vital.
HEART FAILURE
The other important outcome of AC is end-stage HF and heart transplantation. While refractory VT storm are rarely the indication for cardiac transplan-tation in AC, refractory end-stage HF is usually the reason for transplanting AC patients24. Survival have been improved once ICD implantation has been ex-tensively used and the number of malignant VA sur-vivors developing HF has substantially increased. It is reasonable to consider asymptomatic AC patients as stage B heart failure patients, monitor development of HF symptoms and manage risk factors25 in order to prevent or delay the onset of HF26. Fewer studies have focused on searching predictors of progression toward end-stage HF. Progression toward ventricular dilatation and dysfunction seems to be a slow pro-cess27 and onset of signs/symptoms might be insidious. Usually patients with AC who receive transplants for HF have prolonged clinical course28. One recent study described HF in the setting of AC and reported pre-dictors of HF29. In this cohort, women had a higher risk of HF independent of the presence of LV invol-vement; lateral precordial T-wave inversions predic-ted HF and particularly symptomatic LV dysfunction. Patients who developed signs and symptoms of HF before presentation had less arrhythmic burden and therefore a milder arrhythmic phenotype. Desmo-somal desmoplakin (DSP) and digenic heterozygosity were associated with LV dysfunction and HF29,30 and nondesmosomal phospholamban mutation27,31 was associated with LV involvement in several studies. It was reported that young age at presentation (Figure1) and LV involvement are associated with need for cardiac transplantation in the setting of AC28,32.
DISEASE PROGRESSION
Initially it was thought that AC was due to a develop-ment defect of RV myocardium but it has been shown that myocardial atrophy is in fact a consequence of progressive myocytes loss that begins after birth and evolves thereafter2. Furthermore, genetic testing and familial screening showed that relatives who present with subclinical phenotype are at risk of developing manifest AC over time33. Electrocardiographic stu-dies34,35 and serial imaging examinations that longitu-dinally followed AC patients reported development of new repolarization/depolarization criteria36 over time (Figure 2) and progressive ventricular dilatation and dysfunction37,38.
Among asymptomatic family members, mutation positive female siblings had the strongest probability to fulfill definite AC diagnosis at last follow-up33. De-velopment of full AC phenotype (defi nite AC diagno-sis) over time in family members has been predicted by symptomatic status at enrollment and abnormal baseline ECG39.
Classically, it has been considered that electrical abnormalities precedes structural abnormalities39. But it has been shown that subtle structural alterati-ons detected through altered RV deformation pattern can occur before electrocardiographic changes40. And the substrate of altered RV deformation pattern is re-duced local contractility and increased local passive stiffness of the myocardium40. Electrocardiographic abnormalities refl ect the interference of fi brofatty tissue with electrical impulse conduction. ECG may have lower accuracy in detecting early abnormalities than deformation parameters from advanced imaging techniques. Indeed, in family members, abnormal RV deformation at baseline predicted disease progres-sion, primarily development of new electric criteria during follow-up41.
For patients diagnosed in the phase of overt disea-se who already fulfi ll the majority of 2010 Revised Task Force Criteria36, disease progression continues toward severe RV dilatation and dysfunction with right HF and further toward a phase of biventricular involve-ment and dysfunction which mimic dilated cardiomyo-pathy2. In a cohort of defi nite AC patients of which two thirds had structural task force criteria from baseline, significant RV structural progression was predicted by depolarization criteria at baseline while signifi cant LV structural progression was predicted by the presence of phospholamban mutation27.
The prognostic significance of development of new repolarization, depolarization or arrhythmic crite-ria over time has not been defi ned. One study have shown an association between significant structural RV progression and occurrence of the first ICD therapy during follow-up27.
In conclusion, recognition of risk markers is im-portant in order to prevent SCD, but current thera-peutic approach do not prevent the development or progression of the disease.
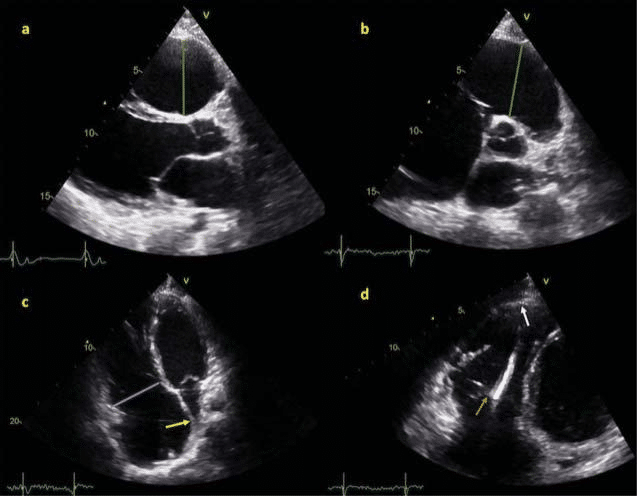
Figure 1. PKP2 mutation positive patient, diagnosed at 17 years old with arrhythmogenic cardiomyopathy, with history of high exercise intensity exposure (10 METs) has been transplanted at 26 years old for end-stage heart failure. Echocardiographic study before heart transplantation shows: Panel a: PLAX view shows increased RVOT diameter 4,9 cm (vertical green line). Panel b: PSAX view shows increased RVOT diameter 5 cm (green line). Panel c: Apical 4 chamber view shows increased basal RV diameter 5,1 cm (blue line) and leftward bulging of atrial septum (yellow arrow). Panel d: RV focused apical 4 chamber view shows thin-walled akinetic apical segment of RV free wall (white arrow) and ICD lead visible in RV cavity (orange arrow).
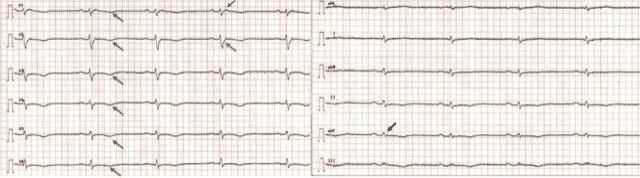
Figure 2. 12-lead electrocardiogram of 47 years old PKP2 mutation positive patient with arrhythmogenic cardiomyopathy shows:
T wave inversion in precordial leads (green arrows) (major repolarization Revised 2010 Task Force criteria); epsilon wave (blue arrow) (major depolarization Revised 2010 Task Force criteria);
terminal activation duration ³ 55 msec (orange arrow) (minor depolarization Revised 2010 Task Force criteria); Fragmented and low-amplitude QRS complexes in limb leads (black arrow).
Abbreviations: PKP2 = plakophilin-2; PLAX = parasternal long axis view; PSAX = parasternal short axis view; RVOT = right ventricle outflow tract; RV = right ventricle; ICD = implantable cardioverter-defibrillator.
Conflict of interest: none declared.
References
1. Haugaa KH, Haland TF, Leren IS, Saberniak J, Edvardsen T. Ar-rhythmogenic right ventricular cardiomyopathy, clinical manifes-tations, and diagnosis. Europace : European pacing, arrhythmias, and cardiac electrophysiology : journal of the working groups on cardiac pacing, arrhythmias, and cardiac cellular electrophysiology of the European Society of Cardiology. 2016;18(7):965-72.
2. Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmo-genic right ventricular cardiomyopathy. Lancet (London, England). 2009;373(9671):1289-300.
3. Sen-Chowdhry S, Syrris P, Ward D, Asimaki A, Sevdalis E, McK-enna WJ. Clinical and genetic characterization of families with ar-rhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation. 2007;115(13):1710-20.
4. Calkins H, Corrado D, Marcus F. Risk Stratifi cation in Arrhyth-mogenic Right Ventricular Cardiomyopathy. Circulation. 2017;136 (21):2068-82.
5. Bhonsale A, James CA, Tichnell C, Murray B, Madhavan S, Phil-ips B, Russell SD, Abraham T, Tandri H, Judge DP, Calkins H. Risk stratifi cation in arrhythmogenic right ventricular dysplasia/cardio-myopathy-associated desmosomal mutation carriers. Circulation Arrhythmia and electrophysiology. 2013;6(3):569-78.
6. Groeneweg JA, Bhonsale A, James CA, te Riele AS, Dooijes D, Tichnell C, Murray B, Wiesfeld AC, Sawant AC, Kassamali B, Ats-ma DE, Volders PG, de Groot NM, de Boer K, Zimmerman SL, Kamel IR, van der Heijden JF, Russell SD, Jan Cramer M, Tedford RJ, Doevendans PA, van Veen TA, Tandri H, Wilde AA, Judge DP, van Tintelen JP, Hauer RN, Calkins H. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Mem-bers. Circulation Cardiovascular genetics. 2015;8(3):437-46.
7. Mazzanti A, Ng K, Faragli A, Maragna R, Chiodaroli E, Orpha-nou N, Monteforte N, Memmi M, Gambelli P, Novelli V, Bloise R, Catalano O, Moro G, Tibollo V, Morini M, Bellazzi R, Napolitano C, Bagnardi V, Priori SG. Arrhythmogenic Right Ventricular Car-diomyopathy: Clinical Course and Predictors of Arrhythmic Risk. Journal of the American College of Cardiology. 2016;68(23):2540-50.
8. Bhonsale A, James CA, Tichnell C, Murray B, Gagarin D, Philips B, Dalal D, Tedford R, Russell SD, Abraham T, Tandri H, Judge DP, Calkins H. Incidence and predictors of implantable cardioverter-defi brillator therapy in patients with arrhythmogenic right ventric-ular dysplasia/cardiomyopathy undergoing implantable cardiovert-er-defi brillator implantation for primary prevention. Journal of the American College of Cardiology. 2011;58(14):1485-96.
9. Corrado D, Calkins H, Link MS, Leoni L, Favale S, Bevilacqua M, Basso C, Ward D, Boriani G, Ricci R, Piccini JP, Dalal D, Santini M, Buja G, Iliceto S, Estes NA, 3rd, Wichter T, McKenna WJ, Thiene G, Marcus FI. Prophylactic implantable defi brillator in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia and no prior ventricular fi brillation or sustained ventricular tachy-cardia. Circulation. 2010;122(12):1144-52.
10. Corrado D, Leoni L, Link MS, Della Bella P, Gaita F, Curnis A, Salerno JU, Igidbashian D, Raviele A, Disertori M, Zanotto G, Ver-lato R, Vergara G, Delise P, Turrini P, Basso C, Naccarella F, Mad-dalena F, Estes NA, 3rd, Buja G, Thiene G. Implantable cardiovert-er-defi brillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2003;108(25):3084-91.
11. Link MS, Laidlaw D, Polonsky B, Zareba W, McNitt S, Gear K, Marcus F, Estes NA, 3rd. Ventricular arrhythmias in the North American multidisciplinary study of ARVC: predictors, character-istics, and treatment. Journal of the American College of Cardiol-ogy. 2014;64(2):119-25.
12. Orgeron GM, James CA, Te Riele A, Tichnell C, Murray B, Bhon-sale A, Kamel IR, Zimmerman SL, Judge DP, Crosson J, Tandri H, Calkins H. Implantable Cardioverter-Defi brillator Therapy in Ar-rhythmogenic Right Ventricular Dysplasia/Cardiomyopathy: Pre-dictors of Appropriate Therapy, Outcomes, and Complications. Journal of the American Heart Association. 2017;6(6).
13. Hulot JS, Jouven X, Empana JP, Frank R, Fontaine G. Natural his-tory and risk stratifi cation of arrhythmogenic right ventricular dys-plasia/cardiomyopathy. Circulation. 2004;110(14):1879-84.
14. Saguner AM, Medeiros-Domingo A, Schwyzer MA, On CJ, Haegeli LM, Wolber T, Hurlimann D, Steffel J, Krasniqi N, Rueger S, Held L, Luscher TF, Brunckhorst C, Duru F. Usefulness of inducible ventricular tachycardia to predict long-term adverse outcomes in arrhythmogenic right ventricular cardiomyopathy. The American journal of cardiology. 2013;111(2):250-7.
15. Saguner AM, Vecchiati A, Baldinger SH, Rueger S, Medeiros-Do-mingo A, Mueller-Burri AS, Haegeli LM, Biaggi P, Manka R, Luscher TF, Fontaine G, Delacretaz E, Jenni R, Held L, Brunckhorst C, Duru F, Tanner FC. Different prognostic value of functional right ventricular parameters in arrhythmogenic right ventricular cardio-myopathy/dysplasia. Circulation Cardiovascular imaging. 2014;7(2): 230-9.
16. Saguner AM, Ganahl S, Baldinger SH, Kraus A, Medeiros-Domin-go A, Nordbeck S, Saguner AR, Mueller-Burri AS, Haegeli LM, Wolber T, Steffel J, Krasniqi N, Delacretaz E, Luscher TF, Held L, Brunckhorst CB, Duru F. Usefulness of electrocardiographic pa-rameters for risk prediction in arrhythmogenic right ventricular dysplasia. The American journal of cardiology. 2014;113(10):1728-34.
17. Corrado D, Wichter T, Link MS, Hauer RN, Marchlinski FE, An-astasakis A, Bauce B, Basso C, Brunckhorst C, Tsatsopoulou A, Tandri H, Paul M, Schmied C, Pelliccia A, Duru F, Protonotarios N, Estes NM, 3rd, McKenna WJ, Thiene G, Marcus FI, Calkins H. Treatment of Arrhythmogenic Right Ventricular Cardiomyopathy/ Dysplasia: An International Task Force Consensus Statement. Cir-culation. 2015;132(5):441-53.
18. Orgeron GM, Te Riele A, Tichnell C, Wang W, Murray B, Bhon-sale A, Judge DP, Kamel IR, Zimmerman SL, Tandri H, Calkins H, James CA. Performance of the 2015 International Task Force Con-sensus Statement Risk Stratifi cation Algorithm for Implantable Cardioverter-Defi brillator Placement in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Circulation Arrhythmia and electrophysiology. 2018;11(2):e005593.
19. Indik JH. Arrhythmic Risk Stratifi cation for Arrhythmogenic Right Ventricular Cardiomyopathy: Should We Ask Who Is at High Risk or Who Is at Low Risk? Circulation Arrhythmia and electrophysi-ology. 2018;11(2):e006160.
20. Bosman LP, Sammani A, James CA, Cadrin-Tourigny J, Calkins H, van Tintelen JP, Hauer RNW, Asselbergs FW, teRiele A. Predicting arrhythmic risk in arrhythmogenic right ventricular cardiomyopa-thy: A systematic review and meta-analysis. Heart rhythm. 2018.
21. Haugaa KH, Basso C, Badano LP, Bucciarelli-Ducci C, Cardim N, Gaemperli O, Galderisi M, Habib G, Knuuti J, Lancellotti P, McKenna W, Neglia D, Popescu BA, Edvardsen T. Comprehensive multi-modality imaging approach in arrhythmogenic cardiomyop-athy-an expert consensus document of the European Association of Cardiovascular Imaging. European heart journal cardiovascular Imaging. 2017;18(3):237-53.
22. Ginghina CE, R. Cardiomiopatia aritmogena de ventricul drept In Mic Tratat de Cardiologie, Editia a II-a. 2017;Editura Academiei Romane, Bucuresti:455-63.
23. Zorzi A, Rigato I, Bauce B, Pilichou K, Basso C, Thiene G, Iliceto S, Corrado D. Arrhythmogenic Right Ventricular Cardiomyopathy: Risk Stratifi cation and Indications for Defi brillator Therapy. Cur-rent cardiology reports. 2016;18(6):57.
24. DePasquale EC, Cheng RK, Deng MC, Nsair A, McKenna WJ, Fon-arow GC, Jacoby DL. Survival After Heart Transplantation in Pa-tients With Arrhythmogenic Right Ventricular Cardiomyopathy. Journal of cardiac failure. 2017;23(2):107-12.
25. Ionescu AAC, M.; Nicula, A.; Popescu, B.A.; Ginghina, C.; Jurcut, R.O. A case of arrhythmogenic cardiomyopathy – not only a right ventricular disease. . Rom J Cardiology 2016;26(4):485-9 (indexed journal CNCSB+ EBSCO).
26. Konstam MA, Kiernan MS, Bernstein D, Bozkurt B, Jacob M, Ka-pur NK, Kociol RD, Lewis EF, Mehra MR, Pagani FD, Raval AN, Ward C. Evaluation and Management of Right-Sided Heart Failure: A Scientifi c Statement From the American Heart Association. Cir-culation. 2018.
27. Mast TP, James CA, Calkins H, Teske AJ, Tichnell C, Murray B, Loh P, Russell SD, Velthuis BK, Judge DP, Dooijes D, Tedford RJ, van der Heijden JF, Tandri H, Hauer RN, Abraham TP, Doevendans PA, Te Riele AS, Cramer MJ. Evaluation of Structural Progression in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. JAMA cardiology. 2017;2(3):293-302.
28. Tedford RJ, James C, Judge DP, Tichnell C, Murray B, Bhonsale A, Philips B, Abraham T, Dalal D, Halushka MK, Tandri H, Calkins H, Russell SD. Cardiac transplantation in arrhythmogenic right ven-tricular dysplasia/cardiomyopathy. Journal of the American Col-lege of Cardiology. 2012;59(3):289-90.
29. Gilotra NA, Bhonsale A, James CA, Te Riele ASJ, Murray B, Tich-nell C, Sawant A, Ong CS, Judge DP, Russell SD, Calkins H, Ted-ford RJ. Heart Failure Is Common and Under-Recognized in Pa-tients With Arrhythmogenic Right Ventricular Cardiomyopathy/ Dysplasia. Circulation Heart failure. 2017;10(9).
30. Bhonsale A, Groeneweg JA, James CA, Dooijes D, Tichnell C, Jongbloed JD, Murray B, te Riele AS, van den Berg MP, Bikker H, Atsma DE, de Groot NM, Houweling AC, van der Heijden JF, Rus-sell SD, Doevendans PA, van Veen TA, Tandri H, Wilde AA, Judge DP, van Tintelen JP, Calkins H, Hauer RN. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/car-diomyopathy-associated mutation carriers. European heart jour-nal. 2015;36(14):847-55.
31. Groeneweg JA, van der Zwaag PA, Olde Nordkamp LR, Bikker H, Jongbloed JD, Jongbloed R, Wiesfeld AC, Cox MG, van der Hei-jden JF, Atsma DE, de Boer K, Doevendans PA, Vink A, van Veen TA, Dooijes D, van den Berg MP, Wilde AA, van Tintelen JP, Hau-er RN. Arrhythmogenic right ventricular dysplasia/cardiomyopa-thy according to revised 2010 task force criteria with inclusion of non-desmosomal phospholamban mutation carriers. The Ameri-can journal of cardiology. 2013;112(8):1197-206.
32. Gilljam T, Haugaa KH, Jensen HK, Svensson A, Bundgaard H, Han-sen J, Dellgren G, Gustafsson F, Eiskjaer H, Andreassen AK, Sjo-gren J, Edvardsen T, Holst AG, Svendsen JH, Platonov PG. Heart transplantation in arrhythmogenic right ventricular cardiomyopa-thy – Experience from the Nordic ARVC Registry. International journal of cardiology. 2018;250:201-6.
33. te Riele AS, James CA, Groeneweg JA, Sawant AC, Kammers K, Murray B, Tichnell C, van der Heijden JF, Judge DP, Dooijes D, van Tintelen JP, Hauer RN, Calkins H, Tandri H. Approach to family screening in arrhythmogenic right ventricular dysplasia/cardiomy-opathy. European heart journal. 2016;37(9):755-63.
34. Piccini JP, Nasir K, Bomma C, Tandri H, Dalal D, Tichnell C, James C, Crosson J, Calkins H. Electrocardiographic fi ndings over time in arrhythmogenic right ventricular dysplasia/cardiomyopathy. The American journal of cardiology. 2005;96(1):122-6.
35. Saguner AM, Ganahl S, Kraus A, Baldinger SH, Akdis D, Saguner AR, Wolber T, Haegeli LM, Steffel J, Krasniqi N, Luscher TF, Tan-ner FC, Brunckhorst C, Duru F. Electrocardiographic features of disease progression in arrhythmogenic right ventricular cardiomy-opathy/dysplasia. BMC cardiovascular disorders. 2015;15:4.
36. Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Protonotarios N, Saffi tz JE, San-born DM, Steinberg JS, Tandri H, Thiene G, Towbin JA, Tsatsopou-lou A, Wichter T, Zareba W. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modifi cation of the Task Force Criteria. European heart journal. 2010;31(7):806-14.
37. Nava A, Bauce B, Basso C, Muriago M, Rampazzo A, Villanova C, Daliento L, Buja G, Corrado D, Danieli GA, Thiene G. Clinical profi le and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. Journal of the American College of Cardiology. 2000;36(7):2226-33.
38. Aneq MA, Lindstrom L, Fluur C, Nylander E. Long-term follow-up in arrhythmogenic right ventricular cardiomyopathy using tissue Doppler imaging. Scandinavian cardiovascular journal: SCJ. 2008; 42(6):368-74.
39. te Riele AS, James CA, Rastegar N, Bhonsale A, Murray B, Tich-nell C, Judge DP, Bluemke DA, Zimmerman SL, Kamel IR, Calkins H, Tandri H. Yield of serial evaluation in at-risk family members of patients with ARVD/C. Journal of the American College of Cardi-ology. 2014;64(3):293-301.
40. Mast TP, Teske AJ, Walmsley J, van der Heijden JF, van Es R, Prinzen FW, Delhaas T, van Veen TA, Loh P, Doevendans PA, Cramer MJ, Lumens J. Right Ventricular Imaging and Computer Simulation for Electromechanical Substrate Characterization in Arrhythmogenic Right Ventricular Cardiomyopathy. Journal of the American Col-lege of Cardiology. 2016;68(20):2185-97.
41. Mast TP, Taha K, Cramer MJ, Lumens J, van der Heijden JF, Bouma BJ, van den Berg MP, Asselbergs FW, Doevendans PA, Teske AJ. The Prognostic Value of Right Ventricular Deformation Imaging in Early Arrhythmogenic Right Ventricular Cardiomyopathy. JACC Cardiovascular imaging. 2018.
 This work is licensed under a
This work is licensed under a