L. Lucaci
„Prof. Dr. George Georgescu” Institute of Cardiovascular Diseases, Iaşi
Laurenţiu Lucaci MD, „Prof. Dr. George Georgescu” Institute of Cardiovascular Diseases, Iaşi, Bulevardul 50 Carol I Boulevard, Postal code: 700503, Iaşi, Tel./fax 0232 219 270, 0232 267 602. E-mail: laurentiulucaci@yahoo.com.
Article received on the 1st of October 2012. Article accepted on the 15th of February 2013.
Abstract: The long QT syndrome is a potentially lethal electric disorder of the heart, characterized by a transient or permanent prolongation of the QT interval, carrying a risk for syncope, due to torsade de pointes and / or for sudden cardiac death, secondary to ventricular fibrillation. It comprises a congenital form and an acquired one. The congenital form has a genetic background, representing one of the several channelopathies, as an inherited dysfunction of a ion channel or of a regulatory protein is their common denominator.The acquired form has an extrinsic cause, usually a QT prolonging drug, an electrolyte imbalance or a bradyarrhythmia, possibly occuring in individuals with low penetrance of the congenital type. The genetic classification, the mechanisms for arrhythmogenesis, as well as the principles of management, tailored to the categories at risk, are presented.
Keywords: long QT, congenital, acquired, syncope, sudden death
Rezumat: Sindromul QT lung se caracterizează prin alungirea temporară sau permanentă a intervalului electrocardiografic QT, asociind risc de sincopă, secundară torsadei vârfurilor şi / sau de moarte cardiacă subită, secundară fibrilaţiei ventriculare. Există o formă congenitală şi una dobândită a sindromului. Forma congenitală reprezintă una dintre aritmiile de origine genetică, datorate disfuncţiei înnăscute a unui canal ionic din membrana celulei miocardice, mai rar a unei proteine reglatoare şi numite astfel canalopatii. Forma dobândită are o mulţime de cauze extrinseci, de obicei un medicament cu potenţial alungitor al QT, o diselectrolitemie sau o bradiaritmie. Sunt prezentate clasificarea genetică actuală a sindromului congenital, mecanismele aritmogenezei, precum şi principiile de tratament, adaptate stratificării riscului.
Cuvinte-cheie: QT lung, congenital, dobândit, sincopă, moarte subită
INTRODUCTION
The long QT syndrome (LQTS) consists of a transient or a permanent prolongation of the QT interval above the upper limit of normal, corrected for the heart rate, age and gender, predisposing to syncope due to torsade de pointes (TdP) type of ventricular tachycardia (VT) and / or to sudden cardiac death, secondary to ventricular fibrillation (VF)1-3. The LQTS has two forms: a congenital and an acquired one. The prevalence of the congenital form is estimated between 1/2000 and 1/3000 live births4,5. The acquired form is more common than the inherited one.
LQTS as a cause of sudden cardiac death
Most sudden deaths arise in connection with a structural defect of the heart, be it an ischaemic (80% of cases), a non-ischaemic or an acute mechanical one (aortic dissection, acute massive pulmonary embolism, interventricular septum rupture, blunt chest trauma). In a minority of cases (5-15%), including most of the cases of LQTS6-9 no morphologic abnormality of the heart can be found (Table 1).
Table 1. Etiology of sudden death in an apparently normal heart (7-10)
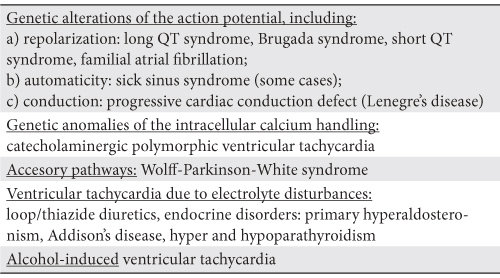
Congenital LQTS inside the genetic anomalies of the ion channels
So far, there is a spectrum of inherited arrhythmia syndromes arising from genetic defects in structures involved in the genesis of action potentials (AP), including congenital LQTS, Brugada syndrome, short QT syndrome (SQTS), catecholaminergic polymorphic ventricular tachycardia (CPVT), idiopathic VF, familial atrial fibrillation (AF) and at least some cases of the sick sinus sindrome (SSS) and of the progressive cardiac conduction defect (PCCD)3,11. Because a ion channel is usually the affected target, these diseases represent the so-called channelopathies11,12, as a group (Figure 1). Either the increased efflux or the decreased influx of positive ions across the cell membrane, through defective channels within at least one myocardial wall layer, bring about short AP syndromes, as for example SQTS and Brugada syndrome. On the other hand, either the increased influx, or the decreased efflux of positive ion, again through defective channels, within at least one myocardial layer, give rise to long AP syndromes, including most of the types of LQTS13. A relationship between the traditional description and the current genetic classification of LQTS has been revealed. Accordingly, the autosomal dominant forms of the genetic types 1, 2, 3, 5 and 6 represent altogether the Romano-Ward syndrome, whereas the autosomal recessive forms of the failing subunits in the slow outward rectifier potassium channel IKs merge into the Jervell and Lange-Nielsen syndrome3,12,14,15.
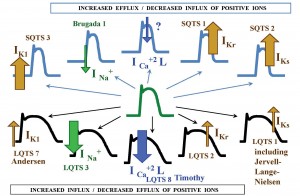
Figure 1. Congenital LQTS as a genetic channelopathy. LQTS = long QT syndrome; SQTS = short syndrome; IK1 = inward rectifier potassium channel; INa+ = sodium channel; ICa+2 L = L-type calcium channel; IKr = rapid component of the delayed outward rectifier channel; IKs = slow component of the delayed outward rectifier channel.
The genetic classification of congenital LQTS and the relationship to the risk profile
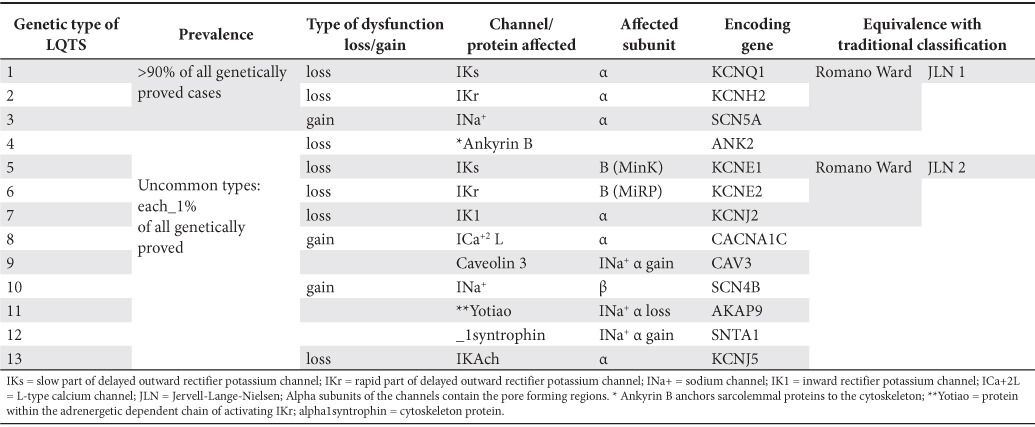
As yet, a number of 13 types of congenital LQTS have been characterized (Table 2). Together, they stand for 75-80% of all patients diagnosed with congenital LQTS. However, the first three ones discovered and therefore allocated as LQTS 1, LQTS 2 and LQTS 3 still remain the most common ones, comprising as a whole 95% of those with a genetic label and at least 70% of all congenital LQTS patients5,16. Most of the cases are the outcome of a direct change of the function (either a loss of function of the repolarizing channels, or a gain of function of the depolarizing ones), secondary to a mutation inside the gene coding for the synthesis of the channel subunit. The remaining cases usually derive from an indirect change of the ion channel function, due to a genetic alteration of a regulatory protein5. There is not just one, but several possible mutations for each type of congenital LQTS (over 700 mutations for all types of LQTS are described by now)5. Every one of them encodes for a region of the ion channel subunit or of the regulatory protein, resulting in different levels of infringement upon channel function and thus various degrees of clinical severity, even inside each type of LQTS, whatever it may be. The degree of the QT prolongation and its temporal variability, the shape of the T wave, the severity of the symptoms, along with some scarce anatomic features inside or outside the heart eventually result from the type of malfunctioning channel / protein and its mutation, as well as from the pattern of inheritance. For instance, among the most three common types of LQTS, IKs (LQTS 1) mutation has the least likely lethal cardiac event (when occurring) and INa+ (LQTS 3) mutation the most likely lethal one3,17,18. ICa+2L (LQTS 8) mutations are presumably lethal in early life, whereas IK1 (LQTS 7) has usually a lenient course5,13,19. Males bear higher risk than females in childhood whatever LQTS type might be20, and throughout life in LQTS 310, while the opposite is true in all other instances for the first three types of congenital LQTS21,22, as well as in the acquired form, a suppressive effect of the estrogens upon IKr channel being hypothesized23. However, the woman’s risk does not lessen at menopause24. Channel conducting pathway (pore) region mutations in LQTS 2 harm more than non-pore coding ones10,25, especially in men26 and the homozygously affected individuals (e.g. Jervell-Lange-Nielsen) evolve worse than their heterozygous counterparts15. Moreover, in the so-called “concealed” cases (genotype-positive, but with normal corrected QT (QTc) at rest), transmembrane (non-pore) region mutations and the genetic type (LQTS 1 and LQTS 3) still carry a higher risk than that of non-affected persons, even if lower than in case of prolonged QTc27. Compared with baseline prolonged QTc individuals, in concealed cases the mutation itself and the genetic type of the syndrome are more powerful risk factors than the clinical descriptors (for example, female gender)27. The greater the number of distinct mutations in the same patients, the earlier the onset of the clinical picture may be.
Table 2. The genetic classification of the types of congenital long QT syndrome (4,5,13,54)
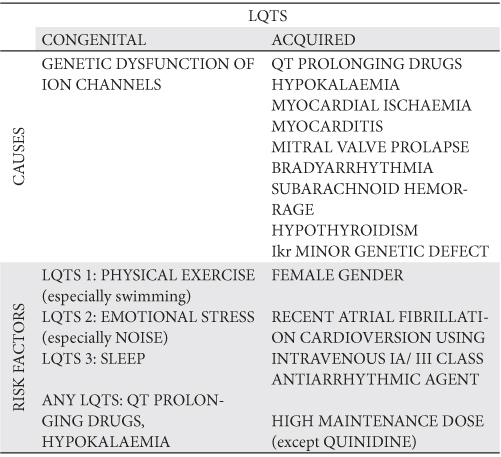
The acquired LQTS
The same IKr channel involved in the pathogenesis of congenital LQTS can be affected by a manifold of drugs or non-pharmacological factors, producing the acquired LQTS13,28,29 (Table 3). Hence, the sinus rhythm electrocardiogram of the acquired syndrome is usually alike to that of the congenital LQTS 2. Unlike the congenital form, a noticeable cause for the acquired syndrome comes into sight. It can be one of the following: a structural cardiac abnormality, bradyarrhythmia, central sympathetic storm (secondary to subarachnoid hemorrhage), electrolyte imbalance (especially hypokalaemia and hypomagnesemia), hypothyroidism, or, more commonly, a QT prolonging drug7,29,30, falling into one of several distinct therapeutic classes, all pertaining to an extensive and currently updating list (www.azcert.org) and blended together because of their ability to block the IKr current29,31, or to inhibit the hepatic metabolism of IKr blockers30. Class IA or III antiarrhythmic drugs are often the culprit agents. Their propensity to prolong QT interval and to increase the TDR is dose-dependent for almost all, except quinidine32. However, the amiodarone is perceived as a relatively safe drug. Its ability to perform a relatively homogenous prolongation of the APs and the attendant Na+ and Ca+2L channels blocking quality result in a lower risk for developing early afterdepolarizations (EADs) and thus for TdP, compared with other antiarrhythmics31,33,34. While safe in most instances, clustering of causes and of risk factors increases dramatically the risk for TdP, and this holds true for amiodarone, too10,28,29,31 (Table 3). As a corollary to the acquired form of LQTS, the discrimination between causes and risk factors is sometimes elusive.
Table 3. Causes and risk factors for LQTS (10,28,29,31)
Issues regarding the measurement and the correction of QT interval
Both the measurement of the QT interval and its adjustment can be done either manually, or by dedicated software built in many present day electrocardiographic machines. Some cautions regarding the manual measurement must be observed, however. A stable isoelectric line and a fairly constant heart rate tracing should be looked for. Choosing the lead with the most clearly defined end of the T wave, from those leads where QT is expected to be the longest (V2 or V3) is recommended35,36. When a TU complex is present, the end of the T wave should be defined by the crossing point between the isoelectric line TP and the tangent line at the steepest downslope of the T wave final portion35. The RR interval used for correction is the RR interval just before the measured QT. The lead chosen as above is kept for subsequent comparisons. Still, one of the most widely used formula to adjust the QT for heart rate is Bazett s formula: QTc = QT / √RR, where QT and RR are measured in seconds1,35,36, so it follows that Bazett QTc is measured in seconds1/2. The fact that QTc is the slope of graph of the square root of RR interval explains the two big shortcomings of the formula, namely the underestimation of QTc (= QT overcorrection) in bradycardia (where the slope of the graph is gentle) and the overestimation of QTc (= QT undercorrection) in tachycardia (where the slope of graph is steep)36,37. Therefore, linear regression formulas are recommended to adjust for rate: QTc = QT + 1.75 × (HR – 60), where QT is measured in miliseconds and HR is the heart rate35,36. Moreover, age and gender must be taken into account, as well. The upper limits of normal for QTc values, as recommended by American Heart Association in 2009 are 0.45 sec for men and 0.46 sec for women, while the lower limit of normal is 0.39 sec for both genders35. To allow for age influence in children, the upper limit of normal of QTc for those younger than 6 months is 0.49 s½ and 0.44 s½ for other age groups, these last values being derived using Bazett formula38. Rate correction becomes useless in case of large RR variability (as for example AF), where the longest observed uncorrected QT should be reported accordingly.
The mechanism of arrhythmogenesis. The ECG diagnosis. “Concealed” cases
Different distributions of potassium channels within ventricular layers (particularly a minimal density of IKs in M-cells)39 contribute to the normal transmural dispersion of AP durations and of the refractory periods (TDR). The M-cell has the longest AP and the subepicardial cell the shortest40,41. The intrinsic failure (seen in congenital LQTS) or the extrinsic impairment of an ion channel in the acquired form prolong the AP of the M-cell and exert variable influences upon the AP durations in the subendocardial and subepicardial layers13. On the one side, one can see the prolongation of QT interval, often associated, but not always, with an increased TDR. On the other side, the long AP of the M-cell allows the Ca+2L channels to be reactivated during the same AP39, promoting excessive Ca+2 storing in the sarcoplasmic reticulum. The ensuing release of intracellular Ca+2 further depolarizes the membrane (by activating Ca+2 dependent chloride current and Na+/ Ca+2 exchange mechanism), giving rise to EADs. Besides further increasing the TDR, an EAD can manifest itself as the ventricular premature beat (VPB) triggering the TdP, if a certain voltage threshold is reached. Thereafter, the augmented TDR favors the perpetuation of the tachycardia by the reentry mechanism30,33,42,43. On the electrocardiogram (Figure 2), the TdP associated with the acquired LQTS and with the congenital LQTS 3 is labeled pause-dependent TdP. Herein it is initiated by a typical sequence, called “short-long-short cycle”32. The first short cycle is represented by the coupling interval of a VPB. The long cycle is the compensatory pause following that VPB, whereas the second short cycle is the coupling interval of the first beat of the tachycardia. In sinus rhythm, QT interval is already long and after the pause it becomes longer32. The twisting pattern of the spikes of QRS complexes around the baseline is explained by a periodic rotation in space of the spiral wave of the tachycardia33. Depending on the amplitude of the EAD and on the value of the TDR, several electrocardiographic scenarios may follow: T wave / TU complex alternance, VPB bigeminism and the TdP. A bidirectional type of VT may also be seen in LQTS 7 (Andersen)11,44. Usually, the TdP is recurrent and self limited, but sometimes (less then 10% of cases) degenerates into VF11,32. TdP in congenital LQTS 1 and 2 may begin without the typical “short-long-short” sequence45. It appears and persists during the sympathetic stimulation in LQTS 1, but only at the beginning of sympathetic release in LQTS 246. Forms of the LQTS where the AP prolongation is quite homogenous have a relatively benign history, with a low prevalence of TdP (LQTS 7 Andersen13, or cases with mild prolongation of QTc under amiodarone as sole risk factor). There has been depicted a characteristic pattern in sinus rhythm for each of the main three types of congenital LQTS, as follows: LQTS 1 has a broad based T wave, LQTS 2 has a low amplitude T wave (sometimes notched, too), whereas LQTS 3 has a narrow based T wave1,2,3,11. The acquired LQTS shows often a pattern similar to that of congenital type 2. Unlike in other types of congenital LQTS, an exaggerated U wave has been described in LQTS 7 (Andersen)10,11,47. Narrow-based T waves (alike those in LQTS 3) and sometimes giant negative T waves in precordial leads are seen in Timothy syndrome48. Aside from the QT prolongation itself and its dispersion in surface leads, the Tpeak-Tend interval in precordial leads (or, more generally, the interval from the peak/nadir of the first component of the T wave to its end) is considered to be a non-invasive indicator of TDR33, while a fragmented QRS is suggested to be a risk indicator for torsade in acquired LQTS, too49. The electrocardiogram of certain cases of congenital LQTS behaves somehow alike to that of the acquired form, as both show a significant variability of QTc over time. It happens especially in heterozygous carriers of IKs mutations in LQTS 1 and LQTS 54. The temporal variability of QTc in congenital LQTS can reach 47 ± 40 ms, including an incidental normal QTc and still carrying a small risk for TdP50. The term “concealed” was coined to label these cases, which may account for 36% of all cases of LQTS 1, 19% of all cases of LQTS 2 and 10% of cases of LQTS 346. The concealed cases fall under the heading of a more general phenomenon of incomplete penetrance of all genetic disorders of the ion channels, while standing for a difficult diagnosis. Persons with the acquired form of LQTS may well be recruited from the bulk of concealed cases. The electrocardiographic diagnosis of concealed LQTS relies upon time (ambulatory Holter recordings) and space follow-up (multiple lead body surface ecg), but provocative tests (exercise stress testing, epinephrine stress) may be needed, as some cases of Brugada syndrome need provocative diagnostic testing (Na+ channel blockers in this case), too4,46. Roughly speaking, the concealed cases of channelopathies are endowed with a risk falling between the risk of spontaneously manifest cases and that of non-carriers, and LQTS does not break this foregoing record6,51.
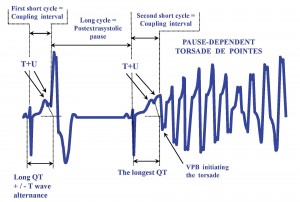
Figure 2. The onset of the pause-dependent torsade de pointes. VPB = ventricular premature beat. Data in references 30,32.
The diagnostic criteria for congenital LQTS presented elsewhere, have been released in 1993, building up a probability diagnosis, founded upon electrocardiographic, personal history and family history elements1,52. Albeit stated before the genetic knowledge breakthrough, they still hold useful, either when genetic diagnosis is out of reach, or as a complementary diagnosis to the genetic work-up done for concealed cases.
Structural abnormalities associated with LQTS
Most cases of congenital LQTS have a normal morphological heart and no extracardiac abnormality. The first historically known abnormality was the sensorineural deafness in Jervell and Lange-Nielsen syndrome14,15,53. Since the beginning of the genetic Odyssey, two other types of LQTS joined the puzzle. One of them is the Andersen-Tawil syndrome (LQTS 7), whose array of features includes a potassium-sensitive periodic paralysis (triggered by exercise or by glucose ingestion), hypertelorism, widened nose base, low-set ears, highly convex palate, small lower jaw, small hands and feet, with syndactyly of the toes5,11,13,47,54,55. The other one is Timothy syndrome (LQTS 8), featuring autism, baldness, low-set ears, small upper jaw, dysmorphic teeth, syndactyly both in fingers and in toes, along with congenital heart defects, such as patent ductus arteriosus, patent foramen ovale, ventricular septal defect, tetralogy of Fallot. Timothy syndrome may include a functional 2/1 atrioventricular block, due to an exceedingly long QT interval5,19,48. Even if the acquired LQTS is fundamentally an electric disorder, a particular anatomic substrate can occasionally be found (myocardial infarction, myocarditis, or a mitral valve prolapse).
Risk stratification
Congenital LQTS: 1) High risk for relapse: patients with personal (not family)10,56 history of aborted cardiac arrest22,57,58, or recent and / or repetitive syncope (especially when first event early in life59,60 and / or on treatment61); 2) Categories at risk for a first cardiac event in life, in the next 5 years (syncope / cardiac arrest / sudden cardiac death) before the age of 40 and without being treated1,21 (Figure 3): 2a) high risk ≥50%: LQTS 1, 2 and 3 with QTc ≥ 500 ms 1/2, apart from the woman with LQTS 3; 2b) low risk <30%: LQTS 1 and 2 with QTc <500 ms1/2, apart from the woman with LQTS 2; 2c) medium risk 30-49%: the LQTS 3 woman with QTc ≥500 ms1/2, the LQTS 2 woman with QTc <500 ms 1/2 and all LQTS 3 patients with QTc <500 ms1/2; 3) risk increased for the woman with LQTS 2 in the first few months after a childbirth, whereas pregnancy is relatively safe62. While being well above the upper limits of normal for both genders, the 500 ms1/2 QTc is a cut-off value for high risk individuals37,50. Acquired LQTS: any clustering of causes and risk factors, especially if symptomatic before.
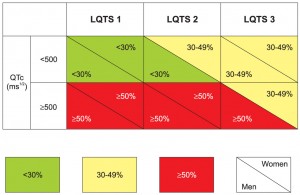
Figure 3. The risk stratification for a first cardiac event in life, before age of 40 and without treatment in congenital LQTS types 1, 2 and 3, depending upon QTc value and gender. High-risk is superior to 50%, medium-risk lies between 30 and 49%, whereas low-risk is inferior to 30%. Data in reference 21.
Principles of management of the LQTS
As not all syncopes in a patient having LQTS are automatically due to TdP, the treatment of any form of LQTS is essentially the prevention and termination of TdP and consequently the prevention of sudden cardiac death. Shortening of QT interval is neither the main goal, nor is always possible. The long-term prevention of TdP in congenital LQTS is attempted by a) avoiding the known risk factors (Table 3), b) safeguard against sympathetic stimulation (betablockers, the mainstay of the treatment1, are mandatory and highly efficient in all cases of LQTS13,20,63, moderately efficient in LQTS 2, including the pregnancy62 and asthma patients64, but are questionable in LQTS 311,65 and in JLN patients15) and c) the possible reduction of the dispersion of refractoriness (by left superior cervico-thoracic sympathectomy, in selected cases66). The long-term prevention of TdP in acquired LQTS is obtained by in-hospital monitoring of the first days of IA / III antiarrhythmic drugs32 and by removal and / or treatment of any identified cause28,32 (Table 3). The prevention of a TdP dependent on a bradyarrhythmia benefits from the bradyarrhythmia treatment10 for any causal LQTS, be it acquired or congenital type 3. Potassium allows the short-term prevention of TdP10, not only in hypokalaemia-induced case, but also in congenital LQTS (here even if no hypokalaemia documented). Stopping TdP in any congenital or acquired case is achieved with intravenous magnesium sulphate and delivery of external electric shock (if TdP episode prolonged or degenerating into VF)32. Moreover, tachyarrhythmia episodes can also be terminated by the implantable cardioverter-defibrillator (ICD) a patient with congenital LQTS has already received67. Some of the aforementioned methods shorten the QTc in sinus rhythm, while others do not (without meaning being less efficient). A synthesis of the available management methods is presented in Figure 4.
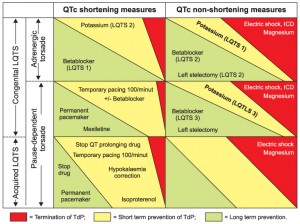
Figure 4. Management of LQTS.
Recommendations for prophylactic management of the congenital LQTS, according to the categories at risk10
All patients must avoid known risk factors (strenuous exercise in LQTS 1, emotional stress and noise in LQTS 2, and any reversible risk factor for the acquired cases) (class I recommendation). Betablockers are recommended (class I) for any patient with electrocardiographic diagnosis and QTc ≥500 ms1/2 (including the pregnant woman) and they are useful (class IIa recommendation) for the patient with QTc <500 ms1/2. The ICD is recommended (class I) for the secondary prevention of sudden cardiac death after an aborted cardiac arrest, it is useful (IIa) in the secondary prevention of both syncope and TdP and it is suggested (IIb) for the asymptomatic patient carrying a high or medium risk (≥30%)68. The ICD must be associated with the betablocker. The left superior cervico-thoracic sympathectomy is suggested (IIb) for unabating symptoms while on betablocker plus ICD.
Conflict of interests: The author declare that no conflict of interest exists.
References
1. Goldenberg I, Moss AJ. Long QT syndrome. J Am Coll Cardiol 2008;51(24):2291-2300.
2. Roden MD. Long QT syndrome. N Engl J Med 2008;358: 69-176.
3. Priori SG, Napolitano C. Genetics of Channelopathies and clinical implications. Hurst’s the heart, 12th edition. Editors: Fuster V, O’Rourke RA, Walsh RA et al. McGraw Hill, New York, 2008,885-897.
4. Brugada R, Campuzano O. The long QT syndrome. Clinical approach to sudden cardiac death syndromes. Editors: Brugada R, Brugada P, Brugada J. Springer Verlag, London, 2010,121-129.
5. Schulze-Bahr E. Long QT syndromes: genetic basis. Long QT syndrome. Cardiac electrophysiology clinics, volume 4, number 1. Editors: Priori SG, Thakur RK, Natale A. Saunders, Philadelphia, 2012,1-16.
6. Napolitano C, Bloise R, Monteforte N, Priori SG. Sudden cardiac death and genetic ion channelopathies: long QT syndrome, brugada syndrome, short QT syndrome, catecholaminergic polymorphic ventricular tachycardia and idiopathic ventricular fibrillation. circulation, 2012;125:2027-2034.
7. Myerburg RJ, Castellanos A. Cardiac arrest and sudden cardiac death. Braunwald’s heart disease: a textbook of cardiovascular medicine, 9th edition. Editors: Bonow RO, Mann DL, Zipes DP et al. Elsevier Saunders, Philadelphia, 2012,845-884.
8. Roşca M, Călin C. Moartea subită şi resuscitarea cardiacă. Mic Tratat de Cardiologie. Editor Ginghină C. Editura Academiei Române, Bucureşti, 2010, 725-735.
9. Le Lorier P, Monahan K. Sudden cardiac death. Cardiology, 2nd edition. Editors. Crawford MH, DiMarco JP, Paulus WP et al. Mosby, Edinburgh, 2004,775-785.
10. ACC / AHA / ESC 2006 Guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology / American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines. Europace 2006;8:746-837.
11. Tester DJ, Ackerman MJ. Genetics of cardiac arrhythmias. Braunwald’s heart disease: a textbook of cardiovascular medicine, 9th edition. Editors: Bonow RO, Mann DL, Zipes DP et al. Elsevier Saunders, Philadelphia, 2012,81-90.
12. Cerrone M, Priori SG. Genetics of sudden death: focus on inherited channelopathies. Eur Heart J 2011;32:2109-2120.
13. Antzelevitch C, Sicouri S. Mechanisms underlying arrhythmogenesis. Long QT syndrome. Long QT syndrome. Cardiac electrophysiology clinics, volume 4, number 1. Editors: Priori SG, Thakur RK, Natale A. Saunders, Philadelphia, 2012,17-27.
14. Popescu V. Sindromul QT prelungit. Revista Română de Pediatrie. 2006;LV(4):379-382.
15. Schwartz PJ, Spazzolini C, Crotti L et al. The Jervell and Lange-Nielsen syndrome: natural history, molecular basis and clinical outcome. Circulation 2006;113:783-790.
16. Zareba W, Goldenberg I, Moss AJ. The role of the implantable cardioverter-defibrillator in the long QT syndrome. Long QT syndrome. Cardiac electrophysiology clinics, volume 4, number 1. Editors: Priori SG, Thakur RK, Natale A. Saunders, Philadelphia, 2012,89-95.
17. Schwartz PJ, Priori SG, Spazzolini C et al. Genotype-phenotype correlation in the long QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 2001;103:89-95.
18. Zareba W, Moss AJ, Schwartz PJ et al. Influence of the genotype on the clinical course of the long QT syndrome. N Engl J Med 1998;339:960-965.
19. Splawski I, Timothy KW, Sharpe LM et al. Cav1.2 Calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004;119:19-31.
20. Goldenberg I, Moss AJ, Paterson DR et al. Risk factors for aborted cardiac arrest and sudden cardiac death in children with long QT syndrome. Circulation 2008;117:2184-2191.
21. Priori SG, Schwartz PJ, Napolitano C et al. Risk stratification in the long QT syndrome. N Engl J Med 2003;348:1866-1874.
22. Hobbs JB, Peterson DR, Moss AJ. Risk of aborted cardiac arrest or sudden cardiac death during adolescence in the long QT syndrome. JAMA 2006;296(10):1249-1254.
23. Ziv O, Kaufman ES. Age and gender modulation of the long QT syndrome phenotype. Long QT syndrome. Cardiac electrophysiology clinics, volume 4, number 1. Editors: Priori SG, Thakur RK, Natale A., Saunders, Philadelphia, 2012,39-51.
24. Buber J, Mathew J, Moss AJ et al. Risk of recurrent cardiac events after onset of menopause in women with congenital LQTS types 1 and 2. Circulation 2011;123 (24):2784-2791.
25. Moss AJ, Zareba W, Kaufman ES et al. Increased risk of arrhythmic events in long QT syndrome with mutations in the pore region of the human ether-a-go-go-related gene potassium channel. Circulation 2002;105(7):794-799.
26. Migdalovich D, Moss AJ, Lopes CM et al. Mutation and gender-specific risk in type 2 LQTS: implications for risk stratification for life-threatening cardiac events in patients with long QT Syndrome. Heart rhythm 2011;8(10):1537-1543.
27. Goldenberg I, Horr S, Moss AJ et al. Risk for life-threatening cardiac events in patients with genotype-confirmed long QT syndrome and normal range corrected QT intervals. J Am Coll Cardiol 2011; 57(1): 51-59.
28. Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med 2004;350:1013-1022.
29. Roden DM, Viswanathan PC. Genetics of acquired long QT syndrome. J Clin Invest 2005;115(8):2025-2032.
30. Surawicz B, Knilans T. Torsade de pointes, ventricular fibrillation and differential diagnosis of ventricular tachycardia. Chou’s electrocardiography in clinical practice, 6th edition. Saunders Elsevier, Philadelphia, 2008,440-455.
31. Shantsila E, Watson T, Lip GYH. Drug induced QT-interval prolongation and proarrhythmic risk in the treatment of atrial arrhythmias. Europace 2007;9(suppl 4):iv37-iv44.
32. Drew B, Ackerman MJ, Funk M et al. Prevention of torsade de pointes in Hospital settings: a scientific statement from the AHA and ACCF. Circulation 2010;121(8):1047-1060.
33. Antzelevitch C. Mechanisms of cardiac arrhythmias and conduction disturbances. Hurst’s the Heart, 12th edition. Editors: Fuster V, O’Rourke RA, Walsh RA et al. McGraw Hill, New York, 2008,913-945.
34. Murray KT, Roden DM. Disorders of cardiac repolarization: the long QT sSyndromes. Cardiology, 2nd edition. Editors. Crawford MH, DiMarco JP, Paulus WP et al. Mosby, Edinburgh, 2004,765-774.
35. Rautaharju PM, Surawicz B, Gettes LS. AHA / ACCF / HRS Recommendations for the standardization and interpretation of the electrocardiogram. Part IV: The ST segment, T and U waves and the QT interval: a scientific statement from the AHA electrocardiography and arrhythmias committee, Council on Clinical Cardiology; the ACCF and the HRS: Endorsed by the International Society for Computerized Electrocardiology. Circulation 2009;119:e241-e250.
36. Mirvis DM, Goldberger AL. Electrocardiography. Braunwald’s heart disease: a textbook of cardiovascular medicine, 9th edition. Editors: Bonow RO, Mann DL, Zipes DP et al. Elsevier Saunders, Philadelphia, 2012,126-167.
37. Vetter VL. Clues or Miscues: how to make the right interpretation and correctly diagnose long QT syndrome – Editorial. Circulation 2007;115:2595-2598.
38. O’Connor M, McDaniel M, Brady WJ. The pediatric electrocardiogram. Part I. Age-related interpretation. Am J Emerg Med 2008;26: 221-228.
39. Rubart M, Zipes DP. Genesis of cardiac arrhythmias. electrophysiologic considerations. Braunwald’s heart disease: a textbook of cardiovascular medicine, 9th edition. Editors: Bonow RO, Mann DL, Zipes DP et al. Elsevier Saunders, Philadelphia, 2012,653-686.
40. Akar FG, Yan GX, Antzelevitch C, Rosenbaum DS. Unique topographical distribution of M cells underlies reentrant mechanism of torsade de pointes in Long QT syndrome. Circulation 2002;105:1247-1253.
41. Antzelevitch C, Shimizu W. Cellular mechanisms underlying the long QT syndrome. Curr Opin Cardiol 2002;17: 43-51.
42. Issa ZF, Miller JM, Zipes DP. Electrophysiological mechanisms of cardiac arrhythmias. Clinical arrhythmology and electrophysiology – a companion to Braunwald’s heart disease, Saunders Elsevier, Philadelphia, 2009,1-26.
43. Ciudin R. Aritmiile cardiace. Mic Tratat de Cardiologie. Editor Ginghină C. Editura Academiei Române, Bucureşti, 2010, 679-710.
44. Pellizzon OA, Kalaizich L, Ptacek LJ, Tristani-Firouzi M, Gonzalez MD. Flecainide suppresses bidirectional ventricular tachycardia and reverses tachycardia-induced cardiomyopathy in Andersen-Tawil syndrome. J Cardiovasc Electrophysiol 2008;19(1):95-97.
45. Noda T, Shimizu W, Satomi K et al. Classification and mechanisms of torsade de pointes initiation in patients with congenital long QT syndrome. Eur Heart J 2004;25:2149-2154.
46. Shimizu W. Diagnostic evaluation of long QT syndrome. Long QT Syndrome. Cardiac electrophysiology clinics, volume 4, number 1. Editors: Priori SG, Thakur RK, Natale A. Saunders, Philadelphia, 2012,29-37.
47. Zhang L, Benson DW, Tristani-Firouzi M et al. Electrocardiographic features in Andersen-Tawil syndrome patients with KCNJ2 mutations: characteristic TU wave pattern pPredict the KCNJ2 phenotype. Circulation 2005;111:2720-2726.
48. Napolitano C, Antzelevitch C. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac voltage-dependent L-Type calcium channel. Circ Res 2011;108:607-618.
49. Moss AJ. Fragmented QRS is associated with torsade de pointes in patients with acquired long QT syndrome. Heart Rhythm 2010; 7(12):1815-1816.
50. Goldenberg I, Mathew J, Moss AJ et al. Corrected QT variability in serial electrocardiograms in long QT syndrome: the importance of the maximum corrected QT for risk stratification. J Am Coll Cardiol 2006;48(5):1047-1052.
51. ESC Guidelines for the diagnosis and management of syncope (version 2009). Eur Heart J 2009;30:2631-2671.
52. Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation 1993;88:782-784.
53. Tyson J, Tranebjaerg L, Bellman S, Wren C, Taylor JFN. IKs and KvLQT1 mutation in either of the two subunits of the slow component of the delayed rectifier potassium channel can cause Jervell and Lange-Nielsen syndrome. Hum Mol Genet 1997;6(12):2179-2185.
54. Moss AJ, Kass RS. Long QT syndrome from channels to cardiac arrhythmias – review series. J Clin Invest 2005;115(8):2018-2024.
55. Tristani-Firouzi M, Jensen JL, Donaldson MR et al. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome). J Clin Invest 2002;110(3):381-388.
56. Kaufman ES, McNitt S, Moss AJ. Risk of death in the long QT syndrome when a sibling has died. Heart Rhythm 2008;5(6):831-836.
57. Liu JF, Jons C, Moss AJ et al. Risk factors for recurrent syncope and subsequent fatal or near fatal events in children and adolescents with long QT syndrome. J Am Coll Cardiol 2011;57(8):941-950.
58. Goldenberg I, Moss AJ, Bradley J et al. Long QT syndrome after age 40. Circulation 2008;117:2192-2201.
59. Spazzolini C, Mullally J, Moss AJ et al. Clinical implications for patients with long QT syndrome who experience a cardiac event during infancy. J Am Coll Cardiol 2009;54(9):832-839.
60. Hurgoiu V. Concepţii actuale în sindromul morţii subite a sugarului. Revista Română de Pediatrie 2007;LVI(4):330-333.
61. Jons C, Moss AJ, Goldenberg I et al. Risk of fatal arrhythmic events in long QT syndrome patients after syncope. J Am Coll Cardiol 2010; 55(8):783-788.
62. Seth R, Moss AJ, McNitt S et al. Long QT syndrome during pregnancy. J Am Coll Cardiol 2007;49(10):1092-1098.
63. Sauer AJ, Moss AJ, McNitt S et al. Long QT syndrome in adults. J Am Coll Cardiol 2007;49(3):329-337.
64. Thottathil P, Acharya J, Moss AJ et al. Risk of cardiac events in patients with asthma and long QT syndrome treated with beta2-agonists. J Am Coll Cardiol 2008;102(7):871-874.
65. Villain E, Denjoy I, Lupoglazoff JM et al. Low incidence of cardiac events with beta-blocking therapy in children with long QT syndrome. Eur Heart J 2004;25(16):1405-1411.
66. Schwartz PJ. The role of the sympathetic nervous system in the long QT syndrome: the long road from pathophysiology to therapy. Long QT syndrome. Cardiac Electrophysiology Clinics, volume 4, number 1. Editors: Priori SG, Thakur RK, Natale A. Saunders, Philadelphia, 2012,75-85.
67. Daubert JP, Zareba W, Rosero SZ et al. Role of implantable cardioverter defibrillator therapy in patients with long QT syndrome. Am Heart J 2007;153(4 suppl):53-58.
68. Zitron E, Thomas D, Katus HA, Becker R. C. Cardioverter defibrillator therapy in the primary and secondary prevention of sudden cardiac death. Appl Cardiopulm Pathophysiol 2012;16:174-191.
 This work is licensed under a
This work is licensed under a