Ovidiu Tica1,3, Otilia- Anca Tica2,3, Elena Rosca1, Larisa Rosan2,3, Vlad Pantea3, Romina Tor2, Mircea Ioan Sandor3, Ioana Ignat-Romanul3, Adrian Hatos4, Mircea-Ioachim Popescu2,3
1 Department of Pathology, County Hospital of Oradea, Romania
2 Clinic of Cardiology, County Hospital of Oradea, Romania
3 Faculty of Medicine and Pharmacy, University of Oradea, Romania
4 Faculty of Social-Humanistic Sciences, University of Oradea, Romania
Abstract: Introduction – Dilated cardiomyopathy (DCM) represents a group of myocardial disorders associated with mechanical and electrical dysfunctions that causes enlargement of both ventricles. Over 50% of primary DCM are familial in nature and the rest have genetic etiology. Secondary DCM may occur during a wide range of diseases. Functionally, DCM is characterized by systolic dysfunction leading finally to a diminished left ventricular ejection fraction. A good correlation has been found between the severity of the condition clinically and the extent and degree of microscopic abnormalities. Methods – The macroscopic features can be best appreciated with a four-chamber view of the heart or by the short axis (bread loafing) slicing technique. Myocardial fragments are harvested, trimmed and kept in fixation solution for minimum 24 hours necessary for microscopic examination. Subsequently, specimens undergoes tissue processing. Results – Gross examination reveals increased heart weight with a globular shape and a decreased tonus. Endocardial fibrous thickening starts over the septal portion of the LV where there is a prominent fi ne reticulation of trabecular muscles. At microscopic level, different degrees of myofibrillar hypertrophy (enlarged, irregularly shaped muscle fibers with hyperchromatic, squared off nuclei) and collagenous fibrosis are detected. Discussions – Familial DCM is proposed to be considered as a form of “cytoskeltopathy”. Interstitial fi brosis contributes to ventricular dysfunction and affects prognosis in patients with DCM. Conclusions – Without cardio-pulmonary transplant, patients with DCM has ominous prognosis with greatly reduced 5-year survival. The most common histologic alteration in DCM patients is the onset of perivascular fibrosis and prolifera-tion of collagen fibers.
Keywords: dilated cardiomyopathy, histopathological examination, myocardial hypertrophy, endocardial fibrosis, cardio-pulmonary transplant
INTRODUCTION
Dilated cardiomyopathy (DCM) brings together a group of primary or secondary myocardial disorders that causes marked enlargement of both ventricles, di-ffuse parietal diskinesia and systolic dysfunction1,2. Although a universal definition of cardiomyopathy is not currently accepted, American Heart Associa-tion (AHA) defined in 2006 cardiomyopathies as „a heterogeneous group of myocardial diseases associ-ated with mechanical and electrical dysfunctions that develops hypertrophy or myocardial dilation having many causes”3. Primary cardiomyopathies (DCM, hypertrophic cardiomyopathy, restrictive cardiomyopathy, arrhyth-mogenic cardiomyopathy, left ventricular noncompac-tion) are defined as inherent myocardial disorders and secondary ones occur in accordance with other disea-ses that affect myocardial function4. It is estimated that in the US, 5 to 6 million patients with heart failure were diagnosed, of which 5-10% had cardiomyopathy as substrate2. Also, DCM prevalen-ce in the US is 36 cases / 100.000 inhabitants, with approximately 10.000 deaths per year1. The estimated European prevalence of DCM is 1: 2500 inhabitants5. Worldwide, DCM is known to be the most com-mon cause of congestive heart failure in the adult po-pulation4. Over 50% of primary DCM are familial in nature (when seen in more than 2 family members), and the rest have genetic etiology (Duchenne muscle dystrophy, Friedreich ataxia, arhrythmogenic cardi-omyopathy). Women who had recently gave birth can develop peripartum primary DCM1,4. Secondary DCM may occur during infectious di-seases, endocrine disorders, inflammatory conditions, lysosomal storage disease, during the usage of toxic substances, in tachyarrhythmia, hypothermia, irradia-tion and others. Secondary toxic DCM can be determined in par-ticular by alcohol and cocaine. Other known cardio– toxic substances are: amphetamines, cobalt, lead, li-thium, mercury, beryllium, metisergide. vitamin B1 and niacin deficiency also affects myocardial function1. Secondary endocrine based DCM is associated with thyroid disease, pheochromocytoma, Cushing’s syndrome, diabetes mellitus and GH hypersecretion. CMD may be induced by inflammatory conditions due to anti – myocardial antibodies: anti -1 receptor antibodies, anti -myosin heavy chain antibodies, anti -myosin heavy chain antibodies, anti-small chain myosin antibodies and anti-troponin antibodies. Diseases that are associated with secondary DCM are: systemic lupus erythematosus, scleroderma, rheumatoid arthri-tis, dermatomyositis, giant cell arteritis (Horton›s di-sease), Kawasaki disease, sarcoidosis. Iatrogenic DCM can be produced by a wide vari-ety of drugs, especially chemotherapy (anthracyclines, transtuzumab, adriamycin) and anti-retroviral medica-tion1. DCM is characterized functional by systolic dys-function, leading to a diminished stroke volumes, ele-vated left ventricular (LV) end-systolic and end-dias-tolic volumes, diminished ejection fraction, increased ventricular chamber dimensions and wall tension, and thinning of LV wall6.
The onset of the disease occurs most frequently in adulthood (20-60 years) with a mean age at presenta-tion at about 50 years7,8. The main symptoms are progressive dyspnea, noc-turnal paroxysmal dyspnea, decreased tolerance to physical activity, orthopnea and occasionally, patients may experience palpitations1. For diagnosis, electrocardiogram (ECG) can orient for etiology of DCM (coronary diseases, LV hyper-trophy, amyloidosis, atrial fi brillation, and others). Thoracic radiography allows the evaluation of cardio-megaly, pleural effusions and pulmonary stasis1. Natremia, Kalemia, the levels of serum urea and creatinine are used to establish prognosis9. The most prominent stratification markers are BNP and NT – ProBNP.
Transthoracic echocardiography is required in all patients with new onset of heart failure and provides valuable data on cavity sizes, myocardial thickness, valvular and both ventricles function. In patients with DCM both ventricles are enlarged with diffuse hypoki-nesia of LV walls and systolic dysfunction. Echocardi-ography has a sensitivity regarding to reduced systolic function around 80% while specificity reaches 100%9. Stress ECG, angiography, multi-slice CT scan, mag-netic resonance imaging can provide additional infor-mation in assessing DCM patients.
Endo – myocardial biopsy is indicated only in acu-te forms of CMD and eventually in post – myocardial treatment monitoring1. Genetic testing is considered useful in familial CMD forms detected by a screening of the first degree rela-tives of the patient1. About 30% of cases have an inherited basis, usually autosomal dominant, due to germline mutation in a gene encoding sarcomeric protein, intermediate fila-ment, nuclear membrane protein, cytoskeletal prote-in, phospholamban or ion channel protein10.
The complex treatment of these patients is based on establishing the main principles: improvement in vi-tal prognosis, symptoms, reduced morbidity and mor-tality. It is desirable to delay or reverse specific heart remodeling in heart failure. Treatment includes non-pharmacological methods, drugs, or implantable medi-cal devices. The only chance of curative treatment of DCM patients is cardiac transplantation in very well-selected cases. The prognostic factors used in the evaluation of DCM patients are: NYHA classes, LV ejection fracti-on, diastolic dysfunction, decrease of right ventricular function, decrease in O2 maximum consumption, re-duction of glomerular filtration rate, decrease in mean blood pressure1. However, the prognosis for patients with persistent DCM who do not undergo cardiac transplantation is poor with an average 5 – year survival rate under 50%11. A good correlation has been found between the se-verity of the condition clinically and the extent and de-gree of microscopic abnormalities, although the some-times focal nature of the changes may be misleading10.
METHODS
Our study included a total 63 deceased patients (from 1st of January 2016 until 31st of December 2016) which 44 were admitted in County Hospital of Oradea and in 19 cases death occurred in the emergency room. In all corpses, pathological autopsy was performed with the approval of Head of the Pathology Department. We excluded trauma related deaths. These procedu-res were carried out in accordance with the proper legislation12.
Subjects were divided into age groups, differentiati-on between sex, environment and main comorbidities. All the data was processed using Microsoft Excel and SPSS Statistics 21.
For the autopsy technique, we used Rokitansky method that implies examining the organs with ma-intaining connections and anatomical relations with nearby structures.
After opening the thorax by removing the sternum, the cervico – thoracic organs (tongue, larynx, bron-chopulmonary piece, the heart with large vessels, es-ophagus, thyroid and the anterior mediastinum) are taken in one piece.
The pericardium is opened using a sharp pointed scissors and the heart is highlighted with the atrial au-ricles and the base (removing pericardial tissue and fat will expose the great vessels).
The macroscopic features can be best appreciated with a four-chamber view of the heart or by the short axis (bread loafing) slicing technique. We sliced the heart at 1 cm intervals in the short axis (horizontal plane) at the apex and continuing to just below the inferior margin of the atrioventricular valve leafl ets. Next, we open the atrial and the remaining portions of the ventricular chambers. After removing the vessels and residual postmortem blood clots the heart is be-ing weighed13.
Myocardial fragments are harvested, trimmed and kept in fixation solution (10% buffered formalin) for minimum 24 hours necessary for microscopic exa-mination. Subsequently, the fragments were being processed using a Leica TP1020 automatic tissue pro-cessor. After processing, the tissue is included in a paraffin block and sectioned at a thickness of 3 to 5 µm using a Leica manual microtome. The sections are displayed on slides, for drying and de – waxing. The slides are stained using Hematoxylin – Eosin, a special stain for collagen fi bers (Masson’s trichrome) and then we mount a coverslip. For examination we used Leica DM 1000 optical microscope and the Nikon E600 po-larized light microscope. The photos were taken using the camera attached to the microscope.
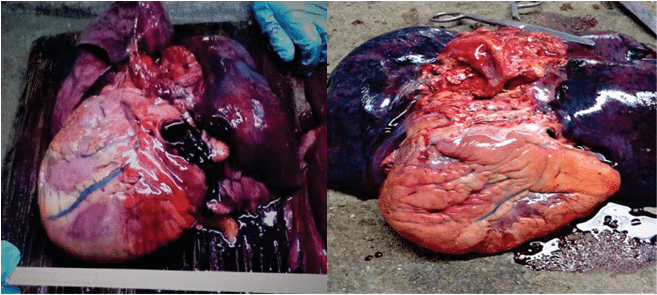
Figure 1. Gross appearances of increased in volume hearts in DCM.
RESULTS
From all 63 cases autopsied with DCM diagnosis, most of them were admitted to Cardiology and Internal Medicine Department (33 corpses). In the Emergency Room 19 patients had DCM in the tanatogenesis of irreversible cardiac arrest. A number of 21 cases had DCM as the main death diagnosis, while cardiomegaly was detected as a comorbidity in 42 patients.
In 6 autopsies performed on female patients, DCM was the main cause of death, despite of 20 body exa-mination, when DCM was an associate diagnosis, in the same gender.
Total number of men with DCM as a primary dia-gnosis was 15, while in 22 autopsies, DCM was found as a comorbidity. In this gender, alcoholic etiology of DCM was suspected in 11 patients that had alcoholic liver cirrhosis and steato – hepatitis without virus im-plication.
In 6 corpses, clear signs of atherosclerosis were not found and in these patients and we excluded ische-mic etiology of DCM. In 17 cases DCM had a valvu-lar background and in 9 patients autopsied, we didn`t found signs of secondary DCM. These cases had a final diagnosis of primary DCM. No peripartum DCM was described in our cohort.
In autopsied patients, mean age was 66,47 years (n=63) with a standard deviation of 13.83 years. Mean age of patients admitted in Cardiology Department was 67.7 years old (n = 10) with a standard deviati-on of 10.15 years that was similar with the age of all admitted patients from our hospital (66,31 years old with a standard deviation of 13.80 years in 44 corp-ses).
By background, we didn’t saw a differentiation between patients from cities or rural sites (30 sub-jects were form urban regions and 33 came to hospital from countryside).
Gross examination reveals increased heart weight with a globular shape and a decreased tonus. On sec-tion, in the initial phases, an adaptive increase in myo-cardial thickness is observed (this muscle hypertrophy is a response to intracavitary pressure). Subsequently, excessive cavity dilatation begins simultaneous with myocardial thinning and may appear as „bovine heart”. Endocardium changes color, becoming greyish with trabecular fibrosis and stiffness. Endocardial fibrous thickening starts over the septal portion of the LV where there is a prominent fi ne reticulation of trabe-cular muscles. The thin myocardium presents diffuse discolorations, fi ne whitish streaks, some of which are branched, with maximal dimensions of up to 5 mm. Intracavitary we can fi nd mural thrombi, especially at apical level. Annular dilated atrioventricular valves ap-pear in the final phases of DCM, which confirms valvu-lar regurgitation diagnosed during life.
At microscopic level, different degrees of myocy-te hypertrophy (enlarged, irregularly shaped muscle fibers with hyperchromatic, squared off nuclei) and collagenous fi brosis are detected. Fibrosis may extend into subendocardial layers, may be diffuse or perivas-cular. Interstitial cellularity is represented by fi brous cells, fi broblasts and possibly lymphocytes (especially
in secondary DCM, postmyocarditis). Some myocardi-al fi bers may exhibit degenerative changes and a peri-nuclear attrition pigment (lipofuscin).
Two important details for differential diagnosis in endomyocardial biopsies are leukocytic infiltrates (is present in about one-half of all patients with myocar-ditis) and the absence of myofibrillar lesions.
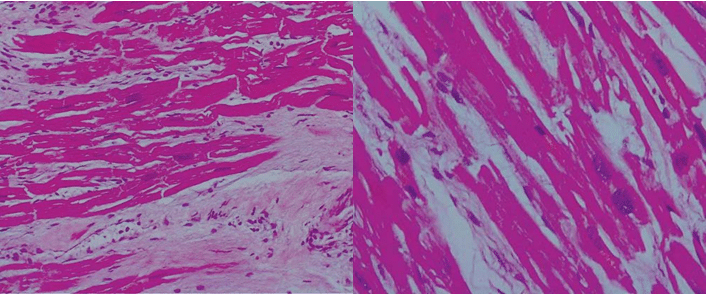
Figure 2. Hematoxylin Eosin stain, hypertrophic myocardial fi bers with „box car” nuclei, fi broblasts and interstitial fibrosis.
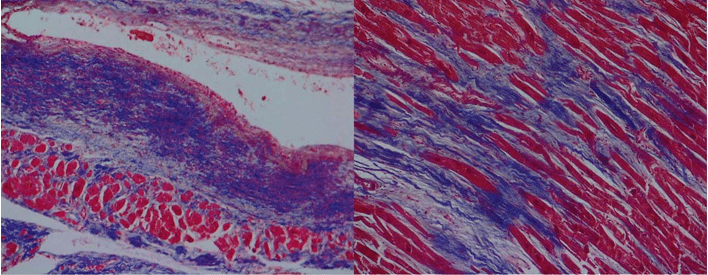
Figure 3. Masson’s trichrome stain showing subendocardial scars and interstitial deposition of collagen fibers.
DISCUSSIONS
Although the onset of DCM occurs with a mean age at presentation at about 50 years, in our study the mean age of death in patients with DCM was 66.47 years old (n = 63)7,8.
Even if we diagnose patients at a younger age, mor-tality rate is higher in elderly patients because of the effectiveness of new cardiac remodeling therapies. The 5-year mortality of patients with non-ischemic CMD is high, with 20% of them having sudden cardiac death. Familial DCM is proposed to be considered as a form of „cytoskeltopathy”14.
A correlation has been found between severity of the clinical signs and the severity of microscopic lesi-ons, although the sometimes focal extent of changes may be misleading10.
Inflammation is an adaptive response to cardi-ac injury, but the role of a prolonged inflammatory response has been well established as maladaptive heart structural alteration. Evidence for cell-mediated immunity includes involvement of pro – inflammatory cytokines and lymphocytic infiltration on biopsies in almost half of patients with idiopathic DCM15.
It is clear that interstitial fibrosis contributes to ventricular dysfunction and affects prognosis in pati-ents with DCM16.
CONCLUSIONS
DCM is known to be the most common cause of con-gestive heart failure. Without cardio-pulmonary trans-plant, patients with DCM has ominous prognosis with greatly reduced 5-year survival.
Early detection and proper treatment may improve the quality of life in these patients.
The most common structural heart alteration in DCM patients is the development of perivascular fi-brosis and proliferation of collagen fibers.
Conflict of interest: none declared.
References
1. Apetrei E. Cardiologie clinica, Editura medicala Callisto, Bucuresti, 2015, 799-814 [37.4].
2. Longo D.L, Fauci A.S. et al, Harrison’s Principles of Internal Medi-cine, Eightteenth Edition, McGraw-Hill Medical, New York, 2012, 1951-1970 [238].
3. Mann D.L., Zipes D.P., Libby P., Bonow R.O., Braunwald E., Braunwald’s heart disease: a textbook of Cardiovascular Medicine, Tenth Edition, Saunders, Philadelphia, 2015, 1551-1573 [65].
4. Suvarna S.K., Cardiac Pathology a guide to current practice, Spring-er, London, 2013, 183-200.
5. Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies: a American Heart Association scientific statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Out-comes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006, 113, 1807-1816.
6. Mills S.E. Sternberg’s Diagnostic Surgical Pathology, Fifth Edition, Lip-pincott Williams & Wilkins, Baltimore, 2010, 1181-1184 [29].
7. Dec G, Fuster V. idiopathic dilated cardiomyopathy. N Engl J Med 1994; 331:1564.
8. General and Systematic Pathology, 4th edition, J.C.E. Underwood, Edit. Churchill and Livingstone, New York, 2007, 320-321 [13].
9. Lindenfeld J, Albert N, Boehmer J, et al., HFSA 2010 Comprehensive Heart Failure Practice Guideline, J Card Fail 2010, 16: e1.
10. Rosai J., Rosai and Ackerman`s Surgical Pathology, Tenth Edition, Elesevier, New York, 2011, 2272-2273 [27].
11. Lilly L.S., Pathophysiology of heart disease: a collaborative project of medical students and faculty, Fifth edition Lippincott Williams & Wilkins, Baltimore, 2011, 245-250 [10].
12. Law 104/2003 towards corpse manipulation with subsequent addi-tions and methodological rules for the application of this law, Moni-torul Oficial al Parlamentului Romaniei, partea I nr. 213 din 25 martie 2014.
13. Finkbeiner W.E., Ursell P.C., Davis R.L., Autopsy pathology: a manu-al and atlas, Saunders Elsevier, Philadelphia, 2009, 42-43 [4].
14. Bowles NE, KR Bowles, JA Towbin, The ‘‘final common pathway’’ hypothesis and inherited cardiovascular disease. The role of cyto-skeletal proteins in dilated cardiomyopathy, Herz, 2000, 25(3):168– 175.
15. Trachtenberg BH, JM Hare, Dilated cardiomyopathy associated with connective tissue disorder, Circ Res 2017 Sep 15;121(7):803-818. doi:10.1161/CIRCRESAHA.117.310221.
16. Soufen HN, VM Salemi, IM Aneas, FJ Ramires, AM Benício, LA Ben-venuti, JE Krieger, C Mady, Collagen content, but not the ratios of collagen type III/I mRNAs, differs among hypertensive, alcoholic, and idiopathic dilated cardiomyopathy, Braz J Med Biol Res, 2008, 41(12):1098–1104.
 This work is licensed under a
This work is licensed under a