Carmen Cristina Olteanu1, Ettore Pedretti2, Giuseppa Privitera2
Articol primit în data de 11 octombrie 2011. Articol acceptat la data 5 decembrie 2011.
1 Spitalul clinic de urgenţă pentru copii “Grigore Alexandrescu”, Bucureşti
2 U.O. Pediatria della Val d’Arda, Piacenza (Italia)
Dr. Cristina Olteanu – Spitalul clinic de urgenţă pentru copii “Grigore Alexandrescu”. Bd. Iancu de Hunedoara Nr. 30-32, 011743. Bucureşti, Sector 1. Tel: +40-21-316.93.66 interior 140
E-mail: c_cristina_olteanu@yahoo.com
Rezumat: Este prezentat cazul unei fetiţe de 6 ani cu dureri musculare, la care investigaţiile paraclinice au condus la diagnosticul de cardiomiopatie dilatativă. Luând prin eliminare majoritatea patologiilor care pot determina această afecţiune cardiacă (de multe ori etichetată idiopatică la copil), s-a identificat cauza disfuncţiei ventriculare şi a durerilor musculare ca fiind deficitul de carnitină. Corectarea tulburării metabolice prin tratament de substituţie a permis restabilirea completă a funcţiei miocardice şi dispariţia simptomatologiei clinice.
Cuvinte cheie: cardiomiopatie dilatativă, carnitină
Abstract: We present a case of a six years old girl with muscular pain. The imaging findings esthablished the diagnosis of dilated cardimyopathy. Cardiomyopathies may be idiopathic or secondary to an underlying systemic disorder (especially in a young population). Taking one by one the majority of etiologies of dilated cardiomyopathy we identified the carnitine deficiency to be the cause of the impaired contractility and left ventricular dilatation and of the muscular pain. The appropriate therapy with carnitine restored the ventricular function and the muscular pain disappeared.
Keywords: dilated cardiomyopathy, carnitine
Introducere
Dilataţia cardiacă cu afectarea funcţiei sistolice este modalitatea uniformă de prezentare a diverselor forme de cardiomiopatie dilatativă (CMD). Factori infecţioşi, ischemici, metabolici, toxici şi ereditari au fost implicaţi în patogeneza acestei afecţiuni, a cărei prevalenţă în populaţie este de 36,5/100.000. La copil, cauza primară a CMD (forma sporadică şi ereditară) nu este identificată în mai mult de 70% din cazuri. Cascada evenimentelor celulare şi moleculare care conduce la instalarea insuficienţei cardiace rămâne de asemenea insuficient cunoscută. Mulţi pacienţi cu CMD idiopatică au o boală silenţioasă clinic în copilărie, dezvoltând simptomele mai târziu, sugerând că există un defect congenital subtil în funcţia miocardică care iniţiază un proces degenerativ gradual1.
Prezentare de caz
Prezentăm cazul unei fetiţe în vârstă de 6 ani, fără antecedente patologice heredocolaterale semnificative, fără antecedente de moarte subită la vârstă tânară în familie, cu o stare bună de sănătate până la momentul internării, cu creştere ponderală în limite normale şi dezvoltare psihomotorie corespunzătoare vârstei, dar care a început să acuze cu 6 luni înainte astenie şi dificultate la mers asociată cu dureri la nivelul musculaturii gambelor şi al musculaturii spinale. Durerile erau intense şi în repaus, producând chiar şi trezirea din somn. Examenul obiectiv la internare: stare de nutriţie bună, paloare cutanată, compensată respirator şi hemodinamic, pulsuri periferice prezente şi simetrice, suflu sistolic 1/6 apical, fără raluri pulmonare, fără hepatomegalie, tonus muscular redus la nivelul membrelor superioare şi inferioare.
Radiografia cardio-pulmonară la internare evidenţia un indice cardio-toracic crescut (Figura 1), iar electrocardiograma (ECG) era modificată: ritm sinusal, ax QRS la stânga, hipertrofie ventriculară stângă (Figura 2).
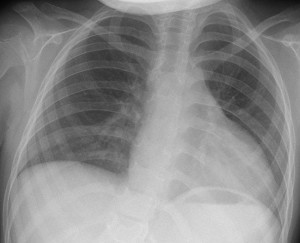
Figura 1. Radiografie cardio-pulmonară în incidenţă postero-anterioară: indice cardio-toracic mărit.
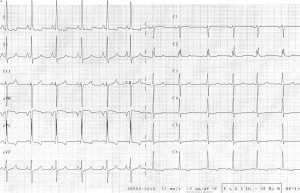
Figura 2. Electrocardiogramă: ritm sinusal 80/minut, ax QRS la stânga, hipertrofie ventriculară stângă.
Ecocardiografia a relevat situs solitus, levocardie, concordanţă atrioventriculară şi ventriculoarterială, întoarcere venoasă pulmonară şi sistemică normală, fără vizualizare de şunt interatrial, sept interventricular intact, valve atrioventriculare şi semilunare cu morfologie normală, regurgitare mitrală uşoară, cavităţi drepte de dimensiuni normale, cavităţi stângi dilatate (diametrul telediastolic ventricul stâng: 47 mm), fracţie de ejecţie globală ventricul stâng redusă (45%), fracţie de scurtare redusă (23%), hipochinezie la nivelul segmentului bazal al septului interventricular şi al peretelui anterior al ventriculului stâng, artere coronare cu origine normală din aortă, presiuni pulmonare estimate normale, fără coarctaţie de aortă, fără flux de canal arterial, pericard indemn. Concluzii: Cardiomiopatie dilatativă (Figura 3).
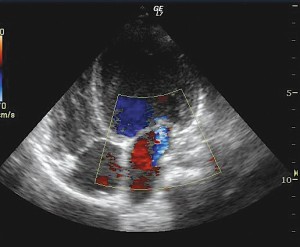
Figura 3. Ecocardiografie transtoracică secţiune apical 4 camere: dilatarea cavităţilor stângi, insuficienţă mitrală uşoară.
Examenele uzuale de laborator au fost negative. CKMB, troponina I, mioglobina au fost negative, homocisteina era în limite normale. Markerii pentru boală de colagen au fost de asemenea negativi, serologia virală pentru Coxsackie, Adenovirus, HIV, Citomegalovirus, Epstein-Barr, Hepatita A, B, C, Herpes virus, Rubeolă, Echo, Parvovirus, virusuri gripale A, B a fost negativă, determinările pentru Toxoplasma, Borellia sau testele endocrinologice (TSH, T3, T4) au fost toate negative. Dozarea nivelului de carnitină plasmatică a evidenţiat un nivel extrem de scăzut (0,7 micromoli/l, valori normale: 21,7-47,3 micromoli/l) cu carnitină urinară dozabilă (determinare calitativă). Înainte de începerea tratamentului de substituţie, suspectând o cauză metabolică a cardiomiopatiei dilatative, ne-am asigurat că pacienta primea o dietă adecvată cu aport proteic crescut şi carne. În aceste condiţii, s-a iniţiat terapia substitutivă cu L-carnitină 300 mg/kg corp/zi (doza uzuală fiind 100-400 mg/kg corp/zi). În timpul administrării carnitinei s-a observat normalizarea valorilor plasmatice de carnitină, chiar depăşind uşor valoarea normală. S-a testat suspendarea administrării carnitinei pentru o săptamână şi a fost reconfirmată valoarea extrem de scăzută sub normal a carnitinei plasmatice. A fost reluată administrarea de L-carnitină 150 mg/kg corp/zi fracţionată în patru prize, fiind continuată şi în prezent cu foarte bună toleranţă. Pe parcursul terapiei cu Carnitină (în doza menţionată anterior), Enalapril (0,3 mg/kg corp/zi, în priză unică) şi Carvedilol (0,4 mg/kg corp/zi în două prize), s-a constatat o ameliorare a datelor ecografice, iar astenia, durerile musculare şi dificultăţile la mers au dispărut. În prezent, la doi ani de la începerea tratamentului de substituţie, nivelul carnitinei plasmatice este 19,5 micromoli/l, iar carnitina urinară este în continuare crescută: 546 micromoli/l (valori normale: 4-29 micromoli/l). Radiografia cardio-pulmonară arată un indice cardio-toracic în limite normale (Figura 4), electrocardiograma s-a normalizat (Figura 5). Ecocardiografia este normală, demonstrând o bună contractilitate miocardică (fracţie de ejecţie globală ventricul stâng: 65%, fracţie de scurtare: 35%), dimensiuni normale ale ventriculului stâng (diametrul telediastolic: 40 mm), fără insuficienţă mitrală (Figura 6, 7).
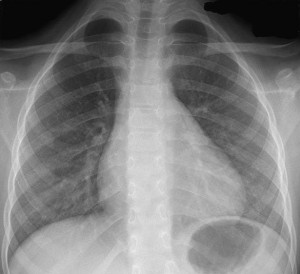
Figura 4. Radiografie cardio-pulmonară în incidenţă postero-anterioară: indice cardio-toracic în limite normale.
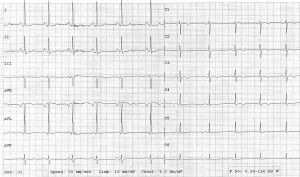
Figura 5. Electrocardiogramă: ritm sinusal 75/minut, ax QRS normal, modificări nespecifice de fază terminală.
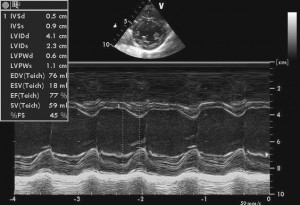
Figura 6. Ecocardiografie transtoracică secţiune parasternal ax scurt: cavităţi stângi de dimensiuni normale.
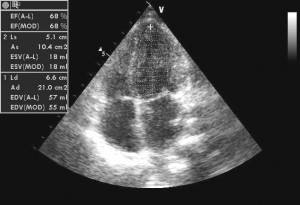
Figura 7. Ecocardiografie transtoracică secţiune apical 4 camere: cavităţi stângi de dimensiuni normale, fracţie de ejecţie ventricul stâng normală.
Discuţii
Oxidarea mitocondrială a acizilor graşi cu lanţ lung este o importantă sursă de energie pentru inimă şi pentru muşchii scheletici în timpul efortului aerobic prelungit. Carnitina (acid beta-hidroxi-gama-trimetilaminobutiric) este un cofactor esenţial pentru transferul acizilor graşi cu lanţ lung prin membrana mitocondrială pentru beta-oxidarea acestora în interiorul mitocondriei. Intervin enzime şi transportori care acumulează carnitina în celulă. OCTN2, transportor membranar plasmatic, transferă carnitina în celulă, aceasta se conjugă cu diferite reziduuri de acil cu ajutorul acil-CoA-sintetazei şi formează acilcarnitina. Acilcarnitina se conjugă cu acizii graşi cu lanţ lung urmând să fie transportată din citosol prin membrana mitocondrială externă cu ajutorul enzimei carnitin-palmitoil-transferază 1 (CPT1). Carnitin-acilcarnitină-translocaza (CACT) transferă acilcarnitina prin membrana mitocondrială internă, iar acizii graşi ajung în matricea mitocondrială unde sunt din nou conjugaţi cu coenzima A cu ajutorul enzimei carnitin palmitoil transferază 2 (CPT2) urmând a fi betaoxidaţi.
Necesarul de carnitină este asigurat prin dietă (proteine de origine animală) şi în mică măsură prin sinteza endogenă din reziduurile de trimetil-lizină generate de catabolismul proteic2. Rata de turnover a carnitinei (300-500 micromoli/ zi) reprezintă sub 1% din depozite. 98% din depozitele de carnitină sunt intracelulare. Carnitina este eliminată prin excreţie urinară după reabsorbţia în proporţie de 98% din cantitatea filtrată, pragul renal pentru carnitină determinând concentraţiile plasmatice şi depozitele totale de carnitină.
Deficitul de carnitină poate fi primitiv sau secundar şi pot fi implicate diferite mecanisme.
La copil deficitul este de obicei primitiv, datorându-se deficitului de transportor membranar plasmatic al carnitinei dependent de sodiu (OCTN2), deficit apărut prin mutaţia genei SLC22A5. Deficitul de carnitină se instalează prin pierdere excesivă renală, fiind afectat procesul de reabsorbţie de la nivelul tubilor renali. Consecinţa este afectarea oxidării acizilor graşi în muşchiul cardiac şi scheletic. În plus, pierderea renală a carnitinei generează nivele plasmatice scăzute, depozite tisulare scăzute şi diminuează captarea hepatică a acesteia prin difuziune pasivă, afectând cetogeneza. Boala este autosomal recesivă şi este numerotată în OMIM ® – Online Mendelian Inheritance in Man ®5 la numărul # 212140, fiind extrem de rară (1/40.000 naşteri). Pacienţii pot avea hipocetoză, hipoglicemie, encefalopatie hepatică, miopatie scheletică sau cardiomiopatie. Această formă răspunde favorabil la suplimentarea aportului de carnitină.
Defectul hepatic al isoformei enzimei CPT1 se prezintă cu atacuri recurente de hipocetoză şi hipoglicemie. Inima şi muşchii care prezintă o formă distinctă de CPT1 nu sunt afectate. Aceşti pacienţi pot avea nivele crescute ale carnitinei plasmatice. Această formă este numerotată în OMIM ® – Online Mendelian Inheritance in Man ®5 la numărul # 255120.
Deficitul de CACT se prezintă de cele mai multe ori în perioada neonatală cu hipoglicemie, hiperamoniemie, cardiomiopatie cu aritmii severe şi moarte subită. Nivelul carnitinei plasmatice este foarte scăzut. Numerotarea în OMIM ® – Online Mendelian Inheritance in Man ®5 este # 212138.
Deficitul de CPT2 se întâlneşte mai frecvent la adult şi se manifestă cu rabdomioliză favorizată de efortul fizic prelungit (OMIM # 255110). Formele mai severe se pot manifesta din perioada neonatală, cu simptomatologie asemănătoare cu cea întâlnită în deficitul de CACT şi pot fi asociate sau nu cu alte anomalii congenitale (forma neonatală letală – OMIM # 608836; forma infantilă – OMIM # 600649).
Tratamentul pentru deficitul de CPT1, CPT2, CACT este dieta săracă în grăsimi şi suplimentarea acesteia cu trigliceride cu lanţ mediu care pot fi metabolizate de mitocondrie independent de carnitină, suplimentarea aportului de carnitină şi evitarea efortului fizic susţinut.
Pe lângă aceste forme, mai este descrisă şi o formă clinică cu manifestări exclusiv miopatice (OMIM # 212160). În aceasta formă cu deficit muscular de carnitină şi miopatie prin încărcare grasă valorile carnitinei serice şi hepatice sunt normale.
Studiile clinice şi pe animale au arătat că o deficienţă moderată pe termen scurt nu are un efect major pe funcţia contractilă miocardică, deşi substratul oxidativ poate fi alterat. Totuşi, după perioade lungi de deficienţă de carnitină se produc alterări în structura inimii, cu afectarea consecutivă a funcţiei contractile. Mecanismul implicat este probabil producţia inadecvată de ATP. De asemenea, deficienţa de carnitină poate produce şi modificări în expresia genică a unor enzime cheie necesare unui metabolism cardiac normal.
Concluzii
În diagnosticul diferenţial al etiologiei CMD, în particular la copil, este importantă determinarea nivelului plasmatic al carnitinei3,4, cu atât mai mult cu cât această tulburare metabolică este corectabilă terapeutic, cu rezultate miraculoase asupra funcţiei miocardice.
Conflict de interese: niciunul.
Bibliografie:
1. Timothy M. Olson, MD, Timothy M. Hofffman, MD, David P. Chan, MD. Dilated congestive cardiomiopathy. In Moss and Adams’ Heart disease in infants, children, and adolescents, Seventh edition. Eds: Hugh d. Allen, MD, ScD( Hon), David J. Driscoll, MD, Robert E. Shaddy, MD, Timothy F. Feltes, MD, Lippincott Williams & Wilkins, 2008, 1195-1207.
2. Stanley CA. Carnitine deficiency disorders in children. Ann N Y Acad Sci. 2004 Nov;1033:42-51.
3. Wang SM, Hou JW, Lin JL. A retrospective epidemiological and etiological study of metabolic disorders in children with cardiomiopathies. Acta Pediatr Taiwan 2006 Mar-Apr; 47(2): 83-7.
4. Gesuete V, Ragni L, Picchio FM. The “big heart” of carnitine. G. Ital Cardiol (Rome). 2010 Sep; 11(9): 703-5.
5. OMIM ® – Online Mendelian Inheritance in Man ®. Johns Hopkins University
 This work is licensed under a
This work is licensed under a