Maria Jalbă1, Sorina Bădeliţă2, D. Coriu2,3, Carmen Ginghină1,3, Ruxandra Jurcuţ1,3*
1 Department of Cardiology, “Prof. Dr. C.C. Iliescu” Institute of Emergency for Cardiovascular Diseases
2 Department of Hematology, Fundeni Clinical Institute
3 University of Medicine and Pharmacy “Carol Davila”, Bucharest
Contact address:
Ruxandra Jurcut, University of Medicine and Pharmacy “Carol Davila”, “Prof. Dr. C.C. Iliescu” Institute of Emergency for Cardiovascular Diseases, Bucharest. E-mail rjurcut@gmail.co
Case presentation
We present the case of a 60 years-old man, presenting with progressive shortness of breath on exertion for the last 2 months. A recent medical evaluation for these symptoms identified important pleural effusion particularly on the left, a pleural tap evacuated 400 ml transsudate, and the cardiac ultrasound showed left ventricular hypertrophy. One year ago he presented at the local hospital with several episodes of paroxysmal atrial fibrillation, for which treatment with amiodarone and oral anticoagulants was started. Two years ago he underwent a coronary angiography, in the setting of atypical angina with an ECG showing R-wave amputation in the right precordial leads, showing normal epicardial coronaries.
At presentation the physical exam shows good general status, a blood pressure of 100/60 mmHg, without postural hypotension, normal lung examination, normal heart sounds, and bilateral lower extremities edema.
The ECG (Figure 1) shows sinus rhythm with low voltage in the limb leads, with a QS pattern in leads DII, DIII, aVF and no R wave progression in V1-V3.
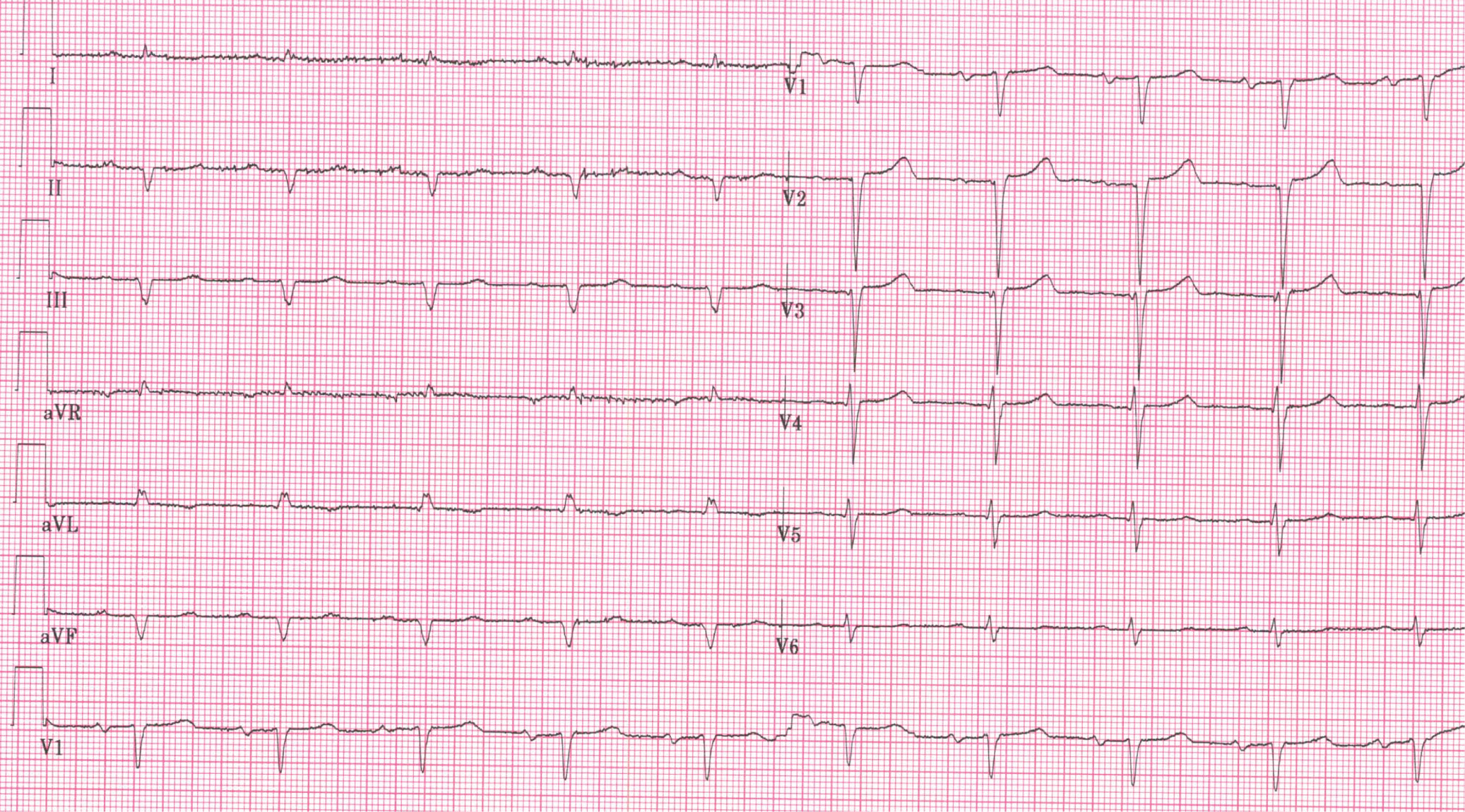
Figure 1. Sinus rhythm with low voltage in the limb leads, with a QS pattern in leads DII, DIII, aVF and no R wave progression in V1-V3.
The laboratory workup shows elevated NT-proBNP (2800 pg/ml) and troponin I (0.079 ng/ml), the rest was unremarkable. Renal function was normal, and there was no proteinuria.
A cardiac ultrasound examination was performed (Figure 2) which showed concentric left ventricle hypertrophy (interventricular septum 18 mm, posterior wall 16 mm), with normal left ventricle systolic function (LVEF 50%) but altered longitudinal function (septal E’ 4 cm/s), restrictive diastolic pattern (E/A 2, E/E’=28) with biatrial dilatation. Global longitudinal myocardial deformation of the left ventricle was found to be severely depressed (GLS=-6.5%) identifying a diffuse subclinical LV dysfunction. The right ventricle was dilated (telediastolic diameter 45 mm), with altered systolic function (TAPSE 13 mm). Moderate functional mitral and tricuspid regurgitation were also present, along with pulmonary hypertension (estimated sPAP=52 mmHg, mPAP=23 mmHg) and a small pericardial effusion.
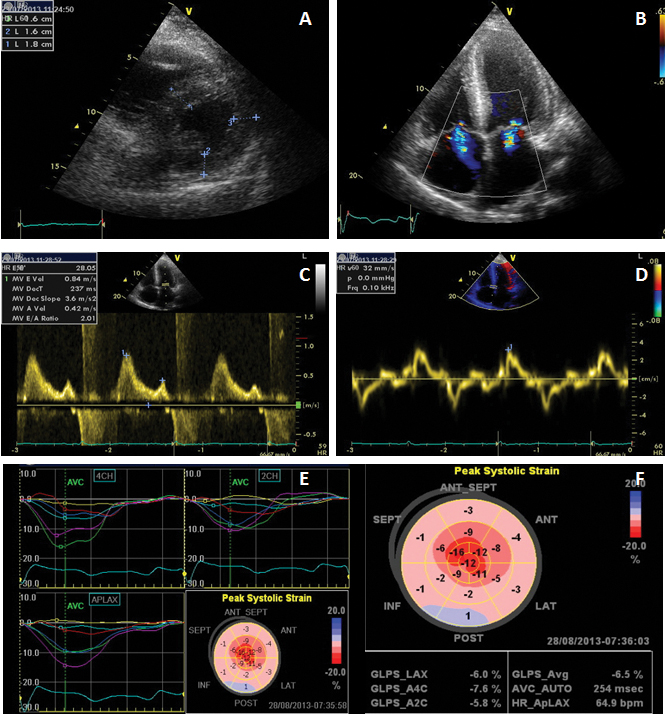
Figure 2. Transthoracic echocardiography examination. A. Parasternal short axis showing concentric LV hypertrophy; B. Color Doppler imaging from apical 4-chambers view showing mitral and tricuspid regurgitations and biatrial enlargement; a sparkling pattern of the interventricular septum can be noted; C. Transmitral flow showing a restrictive pattern; D. Tissue Doppler imaging of the mitral annulus showing decreased longitudinal systolic velocities and E/E’=28; E, F. Automated function imaging of the left ventricle showing diffuse low myocardial deformation with decreased global longitudinal strain (GLS = -6.5%).
Based on the clinical history and examination as well as the above investigations, the diagnosis was of congestive heart failure (functional class NYHA III) due to restrictive cardiomyopathy (CMR). The association between increased left ventricle wall thickness on ultrasound and low QRS voltage on ECG pointed towards a diagnosis of infiltrative cardiomyopathy, with a first line suspicion of cardiac amyloidosis beeing the most frequent cause of CMR.
Cardiac magnetic resonance imaging (MRI) was performed, which confirmed LV hypertrophy with preserved LV global systolic function, and showed diffuse subendocardial late gadolinium enhancement, suggestive of cardiac amyloidosis (Figure 3).
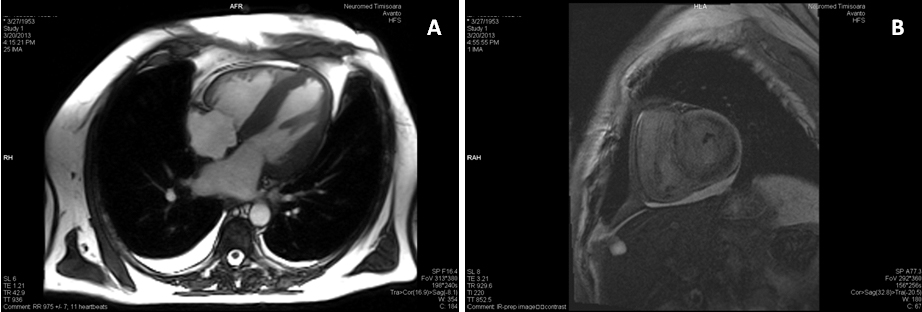
Figure 3. Cardiac MRI. A. Still frame from cine image in 4 chamber-view showing concentric LV hypertrophy, dilated left atrium and small pericardial effusion. B. Delayed sequences after gadolinium administration in short axis shows diffuse intramyocardial delayed enhancement.
In order to obtain a histological confirmation of the disease, an abdominal fat aspirate was performed. The histological preparations stained with Congo red were examined under a microscope with polarized light and amyloid deposits were identified.
The next step was to identify the type of amyloid: AL, formed by the deposition of monoclonal immunoglobulin light chains secreted by a plasmocytes clone in the bone marrow; or familial, where the incriminating protein is a mutant form of transthyretin (so far identified more than 100 mutations); or senile, where the protein is the normal wild-type tranthyretin (a form less likely at the age of 60 years old). Our patient having no history of chronic inflammatory disease, secondary amyloidosis (deposition of serum amyloid A, an acute phase protein) was ruled out.
Serum protein electrophoresis was normal, as well as the level of immunoglobulin; bone marrow biopsy showed only a slight increase of plasmocyte number (5%). These findings are typical for patients which develop AL amyloidosis (primary amyloidosis).
Serum and urine immunofixation were negative. A serum free light chains assay was performed, showing a marked increase of λ light chains, with a κ/λ ratio of 0.09, indicating the presence of a population of plasma cells producing clonal λ free light chains.
To complete the work-up for the extension of the disease, a lower limb electromiography was performed, which showed sensitive and motor polineuropathy.
Thus, a diagnosis of systemic AL amyloidosis (primary amyloidosis) with cardiac and neurological involvement was established. Specific pharmacological treatment was started with dexamethasone, bortezomib and cyclophosphamide.
Discussion
General data on amyloidosis
Amyloidosis represents a group of diseases, characterized by the extracellular deposition of a homogenous material, the amyloid, consisting of fibrils formed by proteins folded in the beta-sheet form. The inertness of amyloid fibrils and their resistance to proteolysis are responsible for their accumulation in tissues and their eventual interference with vital functions. More than 27 types of proteins that can form amyloid deposits have been identified, the most frequent being: immunoglobulin light chains (AL amyloidosis or Primary amyloidosis); familial amyloidosis (mutant transthyretin, lysozyme, fibrinogen, gelsolin); senile systemic amyloidosis (normal wild-type transthyretin); Beta2 – microglobulin amyloidosis (hemodialysis related) and secondary amyloidosis (serum amyloid A). It is a systemic disorder, but the organs most frequently involved are the kidneys, the heart, gastro-intestinal tract, liver, spleen and peripheral and autonomic nervous system1,2.
Cardiac amyloidosis is a myocardial disease resulting from the extracellular amyloid infiltration of the heart. Amyloid deposits occur in the ventricles and atria, as well as perivascularly (particularly in the small vessels) and in the valves. The conduction system may also be involved. The infiltrative process results in biventricular wall thickening with nondilated ventricles, and atrial dilation due to elevation of filling pressures. Cardiac involvment carries a worse prognosis than any other organ involvement3.
Clinical picture
Patients with cardiac amyloidosis present with signs and symptoms of heart failure, with progressive dyspnea, peripheral edema and angina, typical or atypical, due to involvement of the small vessels of the heart by the amyloid deposition. Most patients already have a systemic involvement at the time of cardiac involvement diagnosis. Neurologic symptoms can include autonomic neuropathy that can lead to postural hypotension and syncope, peripheral neuropathy, with sensory disturbances, and carpal tunnel syndrome. The dermatological manifestations of the disease include easy bruising and periorbital purpura, the latter being pathognomonic for AL amyloidosis. Also typical for AL amyloidosis is macroglossia3.
Blood pressure is often low, due to decreased cardiac output in conjunction with early autonomic dysfunction, and in hypertensive patients there is frequently a history of normalization of blood pressure values with the onset of amyloidosis. Pleural effusions and hepatomegaly are frequent, both as a manifestation of congestive heart failure and as a sign of pleural or hepatic involvement of the disease3.
- Electrocardiographic features
- The most frequent ECG abnormality in amyloidosis is low voltage, defined as the presence of QRS
- In all the limb leads <5 mm in height, and due to myocardial amyloid infiltration. A differential diagnosis algorithm in the presence of LV hypertrophy was proposed by Shah et al (Figure 4)4.
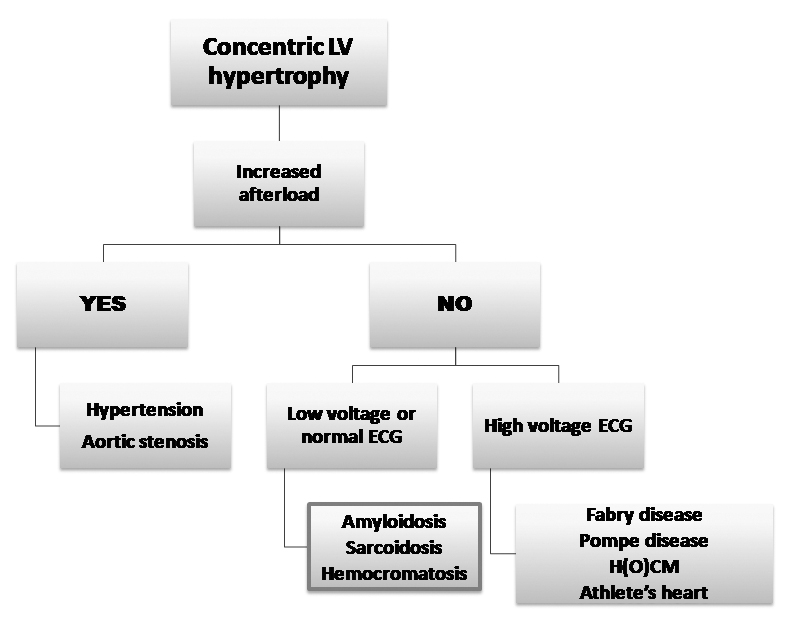
Figure 4. Diagnostic algorithm in left ventricular hypertrophy (modified after 4).
A study identified in 47% of patients with cardiac amyloidosis a pattern of pseudo-infarct, more frequently in the anterior leads, but also inferior or lateral5.
Most patients are in sinus rhythm. Atrial arrhythmias occur in 10% to 15% of patients, the most frequent being atrial fibrillation, which is associated with a very high incidence of thromboembolism. The presence of paroxysmal atrial fibrillation in the patient’s history was probably a first sign of cardiac amyloidosis, either due to atrial involvement or to increased LV filling pressures.
Imaging of cardiac amyloidosis
Echocardiography shows typical features in cardiac amyloidosis: concentric left (and sometimes right) ventricular wall thickening, infiltration of the atrial septum, dilation of the atria, often inceased echogenicity of the valves. The myocardial texture is particular, with increased echogenicity and sometimes granular sparkling (an aspect that is not specific for amyloidosis). The appearance is that of an infiltrative disease, of which amyloidosis is the most frequent cause.
The left ventricular ejection fraction is normal or nearly normal until late in the disease. Diastolic dysfunction progresses with the degree of myocardial infiltration, ranging from impaired relaxation to a restrictive pattern in patients with advanced disease. A small transmitral A wave can be encountered not only in the case of a restrictive pattern but also due to atrial amyloid infiltration and intrinsic atrial dysfunction; in these cases, a normal deceleration time can be associates with a small A wave3. Tissue Doppler imaging can demonstrate the presence of diastolic dysfunction more accurately than transmitral and pulmonary flow, and can detect longitudinal systolic impairment before a change in global systolic function6,7.
Moreover, in a study by Al-Zahrani et al, no diastolic measure approached the accuracy of longitudinal systolic cDMI measurements in identifying ventricular dysfunction in AL patients with normal standard 2D and Doppler examinations, compared to controls8.
Strain and strain rate imaging are even more sensitive than tissue Doppler, allowing the identification of long-axis dysfunction early in the course of the disease, often showing disproportionate impairment of longitudinal contraction despite apparently preserved fractional shortening. Comparing 3 groups of patients (noncardiac amyloid, subclinical cardiac amyloidosis and cardiac amyloidis with clinical signs of congestive heart failure), Koyama et al demonstrated the limitations of standard 2D and Doppler echocardiography for detecting early abnormalities in cardiac function and the value of newer echocardiographic modalities9. The 3 groups of patients all had a mean normal ejection fraction, yet differed in terms of myocardial infiltration and presence of CHF. In contrast to standard echocardiography, strain and SR were able to demonstrate that significant differences in systolic function occur as cardiac amyloid infiltration progresses to CHF. Tissue velocity imaging, a technique that is capable of detecting regional systolic and diastolic function, also failed to differentiate between groups 1 and 2. Our patient was diagnosed in the clinical stage of congestive heart failure; therefore he already associated both low myocardial velocities and strain.
It is important to note that no echocardiographic findings are specific for cardiac amyloidosis, the diagnosis being suggested by a combination of echographic features with low voltage ECG and clinical signs. It should be noted, however, that a decreased ratio of voltage to left ventricular mass is frequent in patients with AL amyloidosis but less so in the hereditary or senile forms, which have frequently normal or increased QRS voltages despite an increase in wall thickness more important than in primary amyloidosis10.
Cardiac magnetic resonance has become an important tool in the diagnosis of cardiac amyloidosis.
It shows a characteristic pattern of global subendocardial late gadolinium enhancement. Gadolinium distributes in the extracellular space, which is expanded by amyloid deposition, leading to signal enhancement; thus, gadolinium follows the histological distribution of amyloid in the myocardium, particularly in the subendocardial layers. Vogelsberg et al showed that this distribution pattern has a 80% sensitivity and 94% specificity for cardiac amyloidosis diagnosis as compared to endomyocardial biopsy in clinical context11. Also, the blood pool is unusually dark, due to high tissue uptake and fast blood pool washout. This leads also to an abnormal myocardial and blood-pool gadolinium kinetics, which proved to have prognostic importance. A study by Maceira et al showed that a low 2 minutes post-gadolinium intramyocardial T1 difference between subepicardium and subendocardium is a valuble tool for predicting an adverse prognosis, by reflecting the increased myocardial amyloid burden. This intramiocardial T1 gradient is a better predictor of survival than free light chain response to chemotherapy12.
Cardiac biomarkers in amyloidosis
Cardiac biomarkers are often elevated in amyloidosis. Troponin increase is due to myonecrosis and small vessel ischemia due to amyloid deposits, and diastolic dysfunction leads to an increased levels of BNP and NT-proBNP. Levels of troponin T and I and NT-proBNP at diagnostic provide prognostic information. A study by Dispenziery et al proposed a staging sistem using troponin and NT-proBNP, using cut-off values of 332 pg/ml for NT-proBNP, and 0,1 ng/ml for cTnI and 0,035 ng/ml for cTnT; patients having neither troponin or NT-proBNP elevation were considered stage I, with a predicted survival of around 27 months; patients with only one elevated marker were considered stage II – predicted survival 11 months, and patients with elevation of both markers stage III, with a predicted survival of 4 months13. Our patient, having only one increased marker, NT proBNP (with Troponin I below the cut-off value) has an intermediate predicted survival.
Diagnostic workup in cardiac amyloidosis
A comparison regarding the sensitivity of different techniques is presented in Table 1, showing the need of at least 2 complementary techniques in order to reach a correct diagnosis.
Table 1. Sensitivity of different diagnostic techniques in the diagnosis of cardiac amyloidosis3,14,15
| Test | Diagnostic elements | Sensitivity (%) |
| ECG | Low QRS voltage Pathologic Q waves |
63-80 60-93 |
| Echocardiography | Interventricular septum hypertrophy Granular sparkling of the miocardium Thickened interatrial septum Valve thickening Biatrial enlargement Pericardic effusion Restrictive transmitral flow pattern |
87 45-87 40-60 65 45 45 35-50 |
| Scintigraphy (Tc 99m pyrophosphate) |
Positive | 23 |
| Cardiac magnetic resonance | Late gadolinium enhancement (mostly global subendocardial deposition) |
80 |
However, the definitive diagnosis of amyloidosis requires a tissue biopsy that identifies amyloid deposits by their apple-green birefringence when stained with Congo red and viewed under polarised light. The most easily accessible are a biopsy from the abdominal fat and from the rectal mucosa. The diagnosis of cardiac amyloidosis can then be established if the echocardiographic or MR appearances are typical, a myocardial biopsy being necessary only if samples from other tissues have been negative.
The next step is establishing the type of amyloid by identifying the precursor protein. AL amiloidosis is the most frequent type, its diagnosis requiring the identification of a plasma cell discrasia. Serum and urine immunofixation should be performed rather than serum and urine electrophoresis because they are much more sensitive for the identification of monoclonal light chains; but even immunofixation fails to identify these monoclonal light chains in 20% of cases16. A serum free light chains assay is even more sensitive, being a usefull tool for diagnosis, prognosis and monitoring of patients with AL amyloidosis. The assay allows the quantification of circulating free lambda and kappa light chains, an abnormal ratio of kappa to lambda being highly suggestive of a population of clonal plasma cells producing free light chains. A bone marrow biopsy is mandatory to asses the percentage of plasma cells (usually 5-10%, the normal being up to 4%) and to excude multiple myeloma, and an imunoperoxidase staining is recommendend to identify wether the plasmocytes secrete kappa or lambda free light chains.
If these are negative, special techniques can be used for the identification of amyloid type: electron microscopy, immunochemistry, aminoacid sequencing and mass spectrometry or genetic testing to rule out familial forms of amyloid2.
Accurate determination of the type of amyloid is essential to establishing the appropriate treatment (for instance chemotherapy and bone marrow transplantation in AL amyloidosis or liver transplantation in transthyretin amyloidosis).
AL amyloidosis is the most frequent amyloidosis type, affecting the heart in 80-90% of cases, with heart failure being the presenting clinical manifestation in about half of these patients. Once congestive heart failure occurs, the median survival is 6 months without treatment3. Studies have proven that cardiac dysfunction in AL amyloidosis is due not only to the myocardial infiltration, but also to intrinsic toxicity of the light chains. The consequences of this mechanism are that myocardial function and symptoms improve in the patients who respond to the hematological treatment, even if on echocardiography the ventricular walls thickness shows no significant improvement17,18.
Management of cardiac amyloidosis
Management of cardiac amyloidosis consists of treatment of the underlying disease along with treatment of the congestive heart failure. The mainstay of the treatment of heart failure in amyloidosis is the use of diuretics; higher doses than anticipated may be required if the albumin level is low as a result of concomitant nephrotic syndrome. Most patients tolerate poorly ACE-inhibitors due to the low blood pressure and orthostatic hypotension. Digoxin is to be used with caution as amyloidosis patients are at increased risk of digitalis toxicity, due to its affinity for amyloid fibrils.
In severe cardiac amyloidosis, the atrium is infiltrated and dysfunctional (with the pressence of atrial electromecanical dissociation) and atrial thrombi may be present even during sinus rhythm3,19,20. Some experts recommend therefore to anticoagulate patients with amyloidosis even if they are in sinus rhythm if there is a small transmitral A wave seen on transthoracic echocardiography (<20 cm/s). Transesophageal echocardiography may be helpful in selected patients in synus rythm, revealing a thombus or spontaneous contrast in the left atrial appendage, or very low atrial appendage Doppler velocities (<40 cm/s)3.
The treatment for AL amyloidosis is administered by hematologists, and aims to destroy the plasma cell clone secreting monoclonal light chains. The standard treatment, similar to that of multiple myeloma, consists of melphalan plus dexamethasone and autologous bone marrow transplantation for eligible patients. Recently, in the treatment of these patients began to be used bortezomib, with good results. A small number of patients have undergone heart transplantation followed by autologous stemm cell transplantation3,16.
Our patient was started on a regimen containing dexamethasone, bortezomib and cyclophosphamide. At 2 months after the start of treatment he is clinically and echocardiographically stable. It should be understood that if the disease is controlled through treatment the prognosis is good. Unfavorable prognosis is given by visceral involvement, especially of the heart. Therefore, heart transplantation should be considered whenever possible, as cardiac amyloidosis can be considered an end-stage heart disease not remediable by more conservative measures.
Conflict of interest: none declared.
References
1. Guan J, Mishra S, Falk RH, Liao R. Current perspectives on cardiac amyloidosis. Am J Physiol Heart Circ Physiol. 2012; 302(3):H544-52.
2. Foerster J. Amyloidosis. In Wintrobe’s Clinical Hematology, 10th ed., 1999 Lippincott Williams & Wilkins (pp 2705-2724).
3. Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation. 2005;112(13):2047-60.
4. Shah KB, Inoue Y, Mehra MR. Amyloidosis and the heart: a comprehensive review. Arch Intern Med. 2006;166(17):1805-13.
5. Murtagh B, Hammill SC, Gertz MA, Kyle RA, Tajik AJ, Grogan M. Electrocardiographic findings in primary systemic amyloidosis and biopsy-proven cardiac involvement. Am J Cardiol. 2005; 95(4):535-7.
6. Koyama J, Ray-Sequin PA, Davidoff R, Falk RH. Usefulness of pulsed tissue Doppler imaging for evaluating systolic and diastolic left ventricular function in patients with AL (primary) amyloidosis. Am J Cardiol. 2002;89(9):1067-71.
7. Koyama J, Davidoff R, Falk RH. Longitudinal myocardial velocity gradient derived from pulsed Doppler tissue imaging in AL amyloidosis: a sensitive indicator of systolic and diastolic dysfunction. J Am Soc Echocardiogr. 2004;17(1):36-44.
8. Al-Zahrani GB, Bellavia D, Pellikka PA, Dispenzieri A, Hayman SR, Oh JK, Miyazaki C, Miller FA Jr. Doppler myocardial imaging compared to standard two-dimensional and Doppler echocardiography for assessment of diastolic function in patients with systemic amyloidosis. J Am Soc Echocardiogr. 2009;22(3):290-8.
9. Koyama J, Ray-Sequin PA, Falk RH. Longitudinal myocardial function assessed by tissue velocity, strain, and strain rate tissue Doppler echocardiography in patients with AL (primary) cardiac amyloidosis. Circulation. 2003;107(19):2446-52.
10. Rapezzi C, Merlini G, Quarta CC, Riva L, Longhi S, Leone O, Salvi F, Ciliberti P, Pastorelli F, Biagini E, Coccolo F, Cooke RM, Bacchi-Reggiani L, Sangiorgi D, Ferlini A, Cavo M, Zamagni E, Fonte ML, Palladini G, Salinaro F, Musca F, Obici L, Branzi A, Perlini S. Systemic
cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120(13):1203-12.
11. Vogelsberg H, Mahrholdt H, Deluigi CC, Yilmaz A, Kispert EM, Greulich S, Klingel K, Kandolf R, Sechtem U. Cardiovascular magnetic resonance in clinically suspected cardiac amyloidosis: noninvasive imaging compared to endomyocardial biopsy. J Am Coll Cardiol. 2008;
51(10):1022-30.
12. Maceira AM, Joshi J, Prasad SK, Moon JC, Perugini E, Harding I, Sheppard MN, Poole-Wilson PA, Hawkins PN, Pennell DJ. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2005;
111(2):186-93.
13. Dispenzieri A, Gertz MA, Kyle RA, Lacy MQ, Burritt MF, Therneau TM, Greipp PR, Witzig TE, Lust JA, Rajkumar SV, Fonseca R, Zeldenrust SR, McGregor CG, Jaffe AS. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol. 2004;22(18):3751-7.
14. Rahman JE, Helou EF, Gelzer-Bell R, et al. Noninvasive diagnosis of biopsy- proven cardiac amyloidosis. J Am Coll Cardiol. 2004;43:410-415
15. Jurcuţ R, Giuşcă S, Enache M, Jurcuţ C, Iacob R, Coriu D, Ginghină C. Ecocardiografia – un nou microscop pentru inimă? Rev Rom Cardio 2008; 23(3):1-5
16. Selvanayagam JB, Hawkins PN, Paul B, Myerson SG, Neubauer S. Evaluation and management of the cardiac amyloidosis. J Am Coll Cardiol. 2007;50(22):2101-10.
17. Liao R, Jain M, Teller P, Connors LH, Ngoy S, Skinner M, Falk RH, Apstein CS. Infusion of light chains from patients with cardiac amyloidosis causes diastolic dysfunction in isolated mouse hearts. Circulation. 2001;104(14):1594-7.
18. Brenner DA, Jain M, Pimentel DR, Wang B, Connors LH, Skinner M, Apstein CS, Liao R. Human amyloidogenic light chains directly impair cardiomyocyte function through an increase in cellular oxidant stress. Circ Res. 2004;94(8):1008-10.
19. Dubrey S, Pollak A, Skinner M, Falk RH. Atrial thrombi occurring during sinus rhythm in cardiac amyloidosis: evidence for atrial electromechanical dissociation. Br Heart J. 1995;74:541–544.
20. Santarone M, Corrado G, Tagliagambe LM, Manzillo GF, Tadeo G, Spata M, Longhi M. Atrial thrombosis in cardiac amyloidosis: diagnostic contribution of transesophageal echocardiography. J Am Soc Echocardiogr. 1999;12:533–536.
 This work is licensed under a
This work is licensed under a