Oliver P Guttmann1, Saidi A Mohiddin2, Perry M Elliott1
1 Inherited Cardiac Diseases Unit, Th e Heart Hospital, University College London, London, UK
2 Department of Cardiology, Th e London Chest Hospital, London, UK
INTRODUCTION
Cardiomyopathies are myocardial disorders that are not explained by abnormal loading conditions and coronary artery disease. They are classified into a number of morphological and functional phenotypes that can be caused by genetic and non-genetic mechanisms. The dominant themes in papers published in 2012–2013 are similar to those reported in Almanac 2011, namely, the use (and interpretation) of genetic testing, development and application of novel non-invasive imaging techniques and use of serum biomarkers for diagnosis and prognosis. An important innovation since the last Almanac is the development of more sophisticated models for predicting adverse clinical events.
HYPERTROPHIC CARDIOMYOPATHY
Cardiac imaging and circulating biomarkers
Hypertrophic cardiomyopathy (HCM) occurs in one in every 500 adults and in most individuals is inherited as an autosomal dominant trait caused by mutations in cardiac sarcomere protein genes and is associated with an increased risk of sudden cardiac death (SCD), progressive ventricular dysfunction and stroke (Figure 1)1-3. Diagnostic tools such as ECG and echocardiography remain fundamental to diagnosis and treatment of HCM but cardiac MRI (CMR) improves diagnostic accuracy and provides additional phenotypic information in patients with established disease (Figure 2)4-7. For example, in one study, CMR identified hypertrophy in about 10% of sarcomere mutation carriers thought to have normal wall thickness by echocardiography 8. Novel CMR sequences, such as T1 mapping, provide quantitative estimates of the myocardial extracellular volume (ECV) (and therefore a surrogate measure of interstitial fibrosis)5 and, in one study, an increase in ECV was reported in individuals with sarcomere mutations but without LV hypertrophy9. These findings suggest that selective use of CMR may be helpful in family screening, particularly when they are other features consistent with HCM, such as ECG abnormalities. The clinical relevance of myocardial scar inferred from abnormal gadolinium enhanced CMR is a recurring subject in the literature. Available data support a relation among late gadolinium enhancement (LGE), representing macroscopic focal myocardial scar, and cardiovascular mortality, heart failure death and all-cause mortality but show only a trend towards an increased risk of SCD10,11. ECV measured by CMR correlates with concentrations of both N-terminal pro-brain natriuretic peptide and serum biomarkers of collagen synthesis providing further evidence that myocardial fibrosis is important early in disease pathogenesis9. Numerous papers have investigated biomarkers as a tool for diagnosis and prognosis and have shown predicting poor outcome in patients with heart failure12. In a study of 772 patients with HCM, brain natriuretic peptide (BNP) was an independent predictor of morbidity in mortality 13. In another study of 183 stable out-patients, plasma NT-proBNP was a predictor of heart failure-related events 14 and was a predictor of heart failure and transplant-related death but not sudden death or inappropriate implantable cardioverter defibrillator (ICD) shocks15. A further study of 183 patients reports elevated serum concentrations of highsensitivity cardiac troponin T to predict adverse outcomes in HCM16.
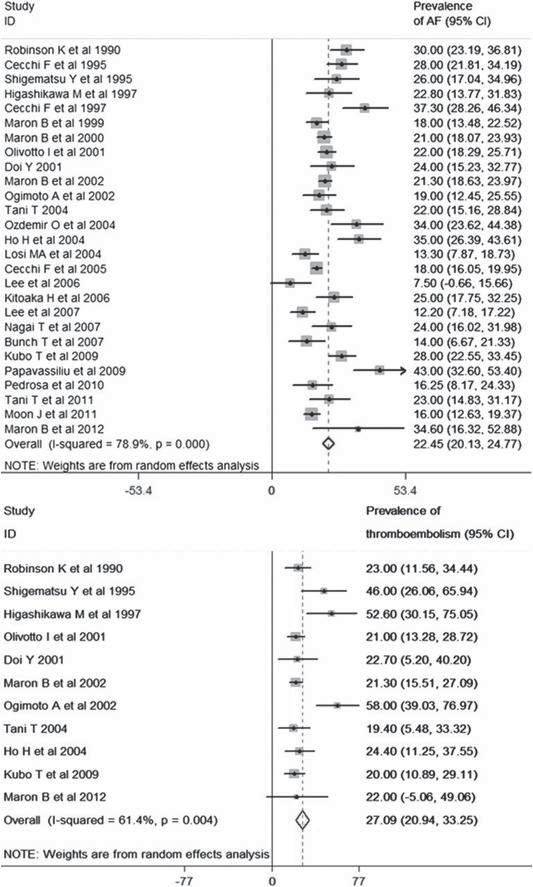
Figure 1. Prevalence of atrial fibrillation and thromboembolism. Guttmann et al3 investigated a population of 7381 patients with hypertrophic cardiomyopathy. A meta-analysis reveals an overall prevalence of atrial fibrillation of 22.45%. The forest plot from random effect meta-analysis shows study-specific prevalence and the pooled (overall) prevalence of AF. The heterogeneity between the study was estimated as I2=78.9% ( p<0.001). The overall prevalence of thromboembolism is 27.09%. The forest plot from random effect meta-analysis shows study-specific prevalence and the pooled (overall) prevalence of thromboembolism in patients with hypertrophic cardiomyopathy and atrial fibrillation. The heterogeneity between the studies was estimated as I2=61.4% (p<0.01).
Treatment strategies
Current management of individuals with HCM focuses on prevention of SCD and stroke, relief of drug-refractory symptoms associated with LVoutflow tract obstruction (LVOTO), and palliation of limiting symptoms caused by systolic or diastolic dysfunction. There have been few developments in therapy since the last Almanac, but early prophylactic β-blocker therapy in physically active patients (NYHA 1 and 2) with provokable LVOTO has been shown to be effective in reducing outflow gradients during physiological exercise17. Another study has confirmed the additive benefit of disopyramide in therapy of symptomatic patients with obstruction resistant to initial therapy with β-blocker or verapamil18. Invasive treatment of LVOTO is recommended for patients with drug-refractory symptoms.
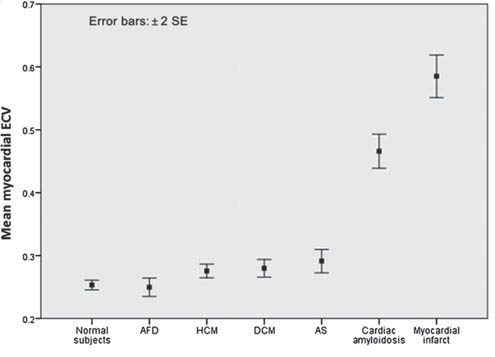
Figure 2. Extracellular volume (ECV) in health and disease: inter-group comparison showing disease-specifi c variability. Sado et al5 measured and assessed the significance of myocardial ECV as a clinical biomarker in health and a number of cardiac diseases. The data are presented as mean±2 SEs.
Several studies have provided new data on septal alcohol ablation (SAA) and LV septal myectomy. Over a follow-up of 5.7 years, survival following SAA in 177 patients was similar to that of patients treated with septal myectomy and a matched control population. Pacemakers were required in 20.3% in patients who underwent SAA compared with 2.3% in the surgical cohort in the 30 days following the procedure19. Similar results following SAA were reported in a study of 470 patients20 in whom 10-year survival (all-cause death rate 1.2%) was 88% compared with 84% in a matched normal population (Figure 3); the same authors also report a reduction in SCD risk factors.
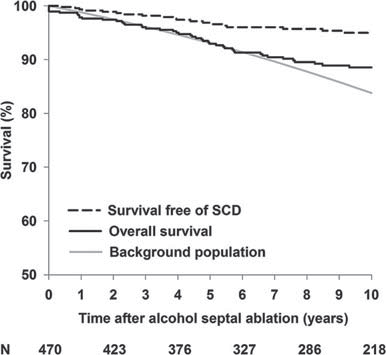
Figure 3. Jensen et al20 investigated overall survival (solid black) and survival free of sudden cardiac death (SCD) including appropriate implantable cardioverter defibrillator discharge and aborted cardiac arrest (dashed black) after alcohol septal ablation in 470 patients with hypertrophic obstructive cardiomyopathy (follow-up 8.4±4 years). Comparison is made with the overall survival of an age- and sex-matched background population (grey). N indicates number of patients at risk at the indicated time.
In a study of 239 patients, septal myectomy was associated with a reduction in syncope and increased survival21. Another study reports a cumulative incidence of HCM-related death of 3.3% at 5 years22. Finally, in 699 patients age and persistent atrial fi brillation were reported to be predictors of poorer outcome in patients undergoing surgical myectomy23. New data have re-examined the efficacy of dual chamber pacing for refractory symptomatic LVO-TO24,25. In a recent Cochrane review, it was noted that all data derive from small studies and that the few randomised trials26,27 concentrate on physiological outcome measures rather than hard clinical endpoints. As a result, they recommend large and high quality trials28.
Prevention of SCD
A recent systematic review and meta-analysis of 27 studies reported an appropriate ICD intervention rate of 3.3% per year with an inappropriate shock rate of 4.8% per annum29 but in a single centre study of 334 patients with HCM, patients still had a significant cardiovascular mortality (predominantly heart failure) and experienced frequent inappropriate shocks and implant complications. These findings suggest that new strategies are required to improve patient selection for ICDs and prevent disease progression in those who receive a device (Figure 4)30. Contemporary risk assessment relies on a small number of readily obtained clinical risk markers to predict SCD and is widely used to guide implantation of ICDs31,32, but recent evidence suggests that this approach distinguishes high and low risk individuals with only limited predictive power33. Additionally, several recently proposed prognostic factors (such as LVOTO and age) are not included in the assessment10,32,34. Recent data have also reinforced the importance of age in risk stratification. In a study of 428 patients above the age of 60 years, 3.7% died secondary to HCM-related causes including embolic stroke, heart failure and transplantation. SCD events occurred in five patients (1.2% or 0.2% per year). The authors conclude that patients with HCM surviving to an age above 60 are at low risk for HCM-related mortality and sudden death 35. It has been suggested that conventional risk prediction algorithms do not apply to paediatric populations 36. In a paediatric cohort, judged clinically to be at high risk, appropriate ICD shocks were delivered in 19% of 224 patients followed for 4.3±3.3 years with an annual rate of 4.5% per year. However, as device-related complications occurred in 41% (especially lead complications and inappropriate shocks) of patients37, more data in paediatric cohorts are required to determine the net benefit of ICD therapy in the young.
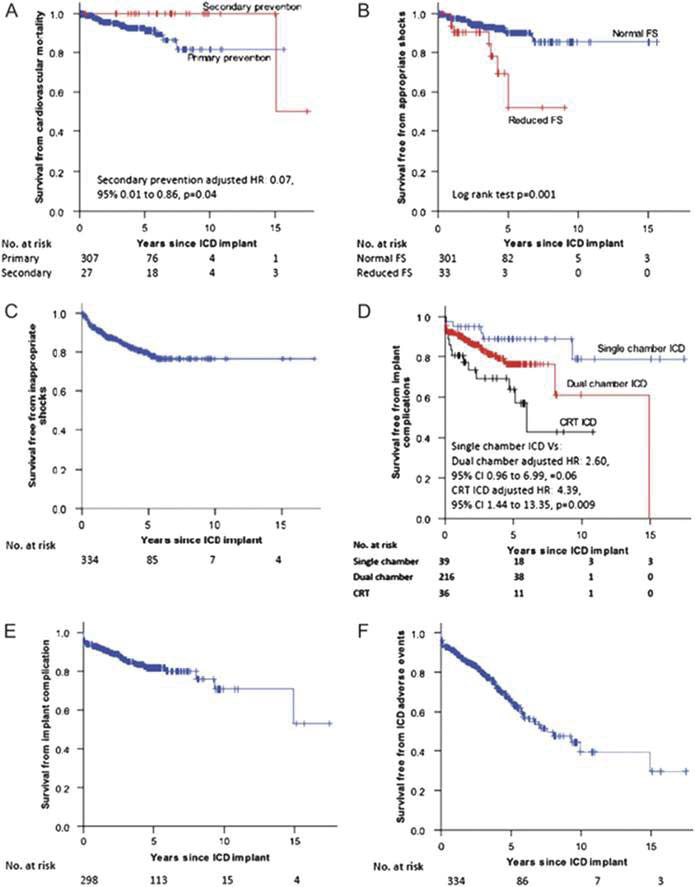
Figure 4. O’Mahony et al30 evaluated 334 patients with hypertrophic cardiomyopathy (HCM) at risk of sudden death treated with ICD. They conclude that HCM patients with an ICD have a significant cardiovascular mortality and are exposed to frequent inappropriate shocks and complications. Kaplan–Meier curves for survival free from cardiovascular mortality end-point (A) stratified according to device implantation for primary or secondary prevention, appropriate shocks (B) stratified according to impaired systolic function, inappropriate shocks (C), implant complications (D) stratifi ed according to device complexity, implant complications in patients with single or dual-chamber ICDs (cardiac resynchronisation therapy patients excluded) (E), and implantable cardioverter defibrillator-related adverse events (inappropriate shocks or implant complications) for the whole cohort (F).
Cost efficacy of family screening
Economic models of genetic testing for the evaluation of families with HCM report that genetic testing is cost-effective when combined with conventional clinical screening38,39. These models are based on the assumption that risk-algorithms from high risk populations can be applied to low risk populations detected through screening and that preventive treatment with ICDs is effective. Additionally, these models and many clinical screening programmes assume a relatively high disease penetrance.In a study comparing clinical screening and predictive genetic testing in children and adolescents, 90 patients with HCM and 361 relatives were followed for 12 years.40 In a group of 12 young mutation carriers without LV hypertrophy, at initial assessment only two developed HCM during the period of the study suggesting an unexpectedly low penetrance during adolescence, a period conventionally associated with the greatest rates of phenotype conversion. Importantly, the two cases were diagnosed at the ages of 26 and 28 years, emphasising the importance of screening beyond adolescence. Conventional clinical screening strategies and the clinical role of genetic testing including its cost-effectiveness will need to be re-evaluated if larger studies show similar findings. Preliminary evidence suggests that long term clinical and genetic screening in children and adults is not associated with major adverse psychological consequences 40. The effect on social and professional aspects has to be evaluated.
Genotype–phenotype relations
The establishment of clinically useful relations between genotype and phenotype remains elusive in HCM. A recent systematic review reported a higher prevalence of family history of HCM and SCD, younger age at presentation, and greater maximal LV in individuals with a sarcomere gene mutation but no difference was reported in clinical characteristics when comparing MYBPC3 and MYH7 mutations. However, these data were limited by the inconsistency of study design and the small size of many study cohorts41. In addition to traditional clinical expression studies, several groups are using human induced pluripotent stem cell (iPSC)-derived cardiac myocytes to study disease pathogenesis. Immunostaining and patch clamping were used to identify disease-specific phenotypes and differences in cardiac drug toxicity between different cell-lines42. The same group used a similar technique to demonstrate that restoration of calcium homeostasis prevented the development of myocyte hypertrophy and electrophysiological abnormalities in pluripotent stem cell-derived cardiomyocytes carrying a MYH7 mutation7.
ARRHYTHMOGENIC RIGHT VENTRICULAR CARDIOMYOPATHY
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is clinically characterised by arrhythmia, SCD and progressive heart failure. Cardiomyocyte loss and replacement by fibrous or fibrofatty tissue are histological hallmarks of the disease. ARVC is caused by mutations in genes that encode constituents of the intercalated disc of cardiomyocytes in a large proportion of patients44. Diagnosis requires integration of data from family members, genetic testing, electrocardiography and imaging techniques45. SCD and treatment of symptomatic arrhythmia and heart failure are the major management challenges.
Clinical diagnosis of ARVC
While evidence suggests that recent modification of proposed diagnostic criteria has improved diagnostic sensitivity and specificity46,47, there is a concern that they remain too sensitive in particular scenarios, most notably athletes and in people of black African ethnic origin, as many structural and electrographic changes considered normal in these groups are also minor diagnostic criteria for ARVC 48,49. Novel methods to detect early phenotypic expression in ARVC have included immunohistochemical and electrophysiological approaches. A report suggested that immunohistochemical demonstration of reduced plakoglobin signal in myocardial biopsies has a sensitivity of 85% and specificity of 57% for ARVC. The authors suggested the test could be used in diagnosis 50, but the performance of the assay in prephenotypic cases—where it is most valuable—has not been assessed. Another group of investigators observed a marked reduction in immunoreactive signal for plakoglobin at cardiac myocyte junctions in patients with sarcoidosis and giant cell myocarditis51. This suggests new disease mechanisms involving desmosomal proteins in granulomatous myocarditis and implicates cytokines in dislocation of plakoglobin from desmosomes and in the development of arrhythmia in ARVC. A recent study reported abnormalities in conduction repolarisation kinetics detected by invasive electrophysiological testing in 10 unrelated individuals and a mouse model with desmoplakin mutations 52. Notably, these abnormalities preceded overt structural changes.
Aetiology
ARVC is inherited as an autosomal dominant trait in up to 50% of cases53 and incomplete penetrance (including age-dependent penetrance) and variable clinical expression are the norm. Over the last year, the genetic heterogeneity of ARVC has been underlined by reports of novel mutations in the genes for phospholamban, desmocollin-2, TMEM43, CTNNA3 (α T catenin) and a gene deletion in plakophilin-2 54-60. Additionally, mutations in genes hitherto associated with other cardiomyopathies have been reported in families and studies of individuals with ARVC. These include the non-desmosomal protein lamin A/C61. The role of other genetic and epigenetic mechanisms on disease expression remains an area of active research 62. Advances in genetics may improve the specificity of diagnostic algorithms in the future but data show that there are many challenges in the interpretation of sequence data. In a study of 427 controls and 93 ARVC probands63, exons and splice acceptor/donor sites of PKP2, DSP, DSG2, DSC2 and TMEM43 were sequenced. Likely pathogenic mutations were identified in 58% of the ARVC cases, but were also found in 16% of the controls. The majority (43%) of the candidate mutations in cases were radical (ie, splice site, nonsense, inframe and frame-shift insertions and deletions) compared with only 0.5% of controls but the frequency of missense mutations was similar in cases (21%) and controls (16%). Other important findings were a higher frequency of candidate variants, in particular missense, in non-Caucasian versus Caucasian controls (19.44% vs 5.83%) and similar numbers of variants in the DSP, DSC2 and TMEM43 genes in the ARVC and control groups. These findings illustrate the conservative approach that must be followed when interpreting genetic variants in ARVC. The use of iPSCs as a model of ARVC was recently described 64. Heterozygous mutant plakophilin-2 iPSC cardiomyocytes demonstrated exaggerated lipogenesis and apoptosis and calcium-handling deficits were detected in homozygous mutants. Greater understanding of these phenomena might lead to development of novel disease-modifying therapeutic strategies in the future.
Management strategies
Once ARVC has been diagnosed, management should include an assessment of SCD risk, indications for drug therapy and need for lifestyle changes. Although anti arrhythmic drugs such amiodarone and sotalol are frequently prescribed to decrease arrhythmic burden44, there is little evidence that these improve survival or alter the natural history of disease. The same is true for treatment of any associated LV systolic impairment with ACE inhibitors and β-blockers. Physical exercise and competitive sport have been reported to increase the risk of sudden death65,66 and are consequently not recommended 67,68. More recently, exercise has been associated with increased disease penetrance and arrhythmic risk in individuals with a desmosomal mutation. Task force criteria were more likely to be met at follow-up and symptoms developed at a younger age in 56 endurance athletes carrying a mutation compared with more sedentary mutation carriers. These athletes also had a decreased lifetime survival free from ventricular tachycardia (VT)/ventricular fibrillation and heart failure 69. These findings corelate with preclinical observations in murine model of ARVC with plakophilin gene defects70. Catheter ablation to treat recurrent VT in ARVC has been associated with high recurrence rates 71,72 but a recent multicentre study with focus on newer ablation strategies assessed recurrence of VT following radiofrequency ablation and effect on burden of VT. The authors report a significant reduction in VT burden and longer survival free of VT following epicardial ablation compared with an endocardial procedure. However, recurrence rates remain considerable with an overall freedom from VTof 47% at 1 year73. These data suggest that catheter ablation may be helpful in a subgroup of patients with incessant or frequent VT refractory to medical therapy. Some data suggest that VT attributable to localised disease is a further potential indication74.
Prevention of sudden death
Current AHA/ACC/ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of SCD recommend ICD implantation in patients with ARVC who have documented sustained VTor ventricular fibrillation and are receiving optimal medical treatment75. A recent literature review investigating outcomes and complications of ICD implantation in ARVC included 610 patients. During the 3.8-year follow-up, the authors reports appropriate ICD interventions at a rate of 9.5% per year with inappropriate interventions (3.7%) and complications including lead malfunction, displacement and infection (20.3% rate of any complication)76. Once again, this underlines the need for appropriate risk stratification to minimise morbidity secondary to ICD-related complications. It is important to point out that the patients analysed in this review received an ICD for primary or secondary prevention. This could at least partially account for the high rate of appropriate interventions. The incremental value and role of CMR in risk stratification were investigated in 69 patients with ARVC-associated mutations (83% with PKP-2 mutations) without prior sustained VT77. Electrical abnormalities were found in 61% of patients of whom 48% also had an abnormal CMR (defined as presence of at least a minor task force criterion). Only a single patient (4%) without electrical abnormalities had an abnormal heart on imaging at baseline. Over a period of 5.8±4.4 years, episodes of sustained VT only occurred in patients with ECG and CMR abnormalities. The authors concluded that among mutation carriers the presence of both electrical and CMR abnormalities identified high risk patients. A similar study on prognosis in patients under evaluation for ARVC reports a positive predictive value of an abnormal CMR in 369 patients who fulfilled at least one minor or major diagnostic criterion for ARVC. The negative predictive value of a normal CMR was 98.8% over a follow-up of 4.3±1.5 years78. The search for biomarkers that allow early diagnosis and risk stratification is an active area of research. For example, low serum concentrations of Bridging integrator 1, a membrane-associated protein, were associated with ventricular arrhythmias and reduced functional status in a small cohort of 24 patients with ARVC 79. A novel strategy to risk prediction in ARVC-associated desmosomal mutation carriers proposes the use of pedigree evaluation, ECG and Holter information80. Phenotypic characteristics were used in stratifying the risk of sustained VT. The investigators included 215 patients over a mean follow-up of 7 years. Patients were risk stratified according to repolarisation and depolarisation abnormalities on ECG. Event-free survival at 5 years was 33% in the high risk group versus 97% in the low risk group.
DILATED CARDIOMYOPATHY
Dilated cardiomyopathy (DCM) is one of the commonest heart muscle diseases in developed countries. It is defined as systolic impairment and LV dilatation in the absence of previous myocardial infarction. Research highlighting the importance of genetics in the aetiology of inherited and apparently acquired forms of DCM has been a prominent feature over the last few years. Standard symptomatic and prognostic heart failure treatments are the mainstay of patient management but more attention has been paid recently to the importance of aetiology in guiding the approach to management.
Genetic subtypes of DCM
A number of studies have examined the natural history of DCM caused by mutations in the lamin A/C gene (LMNA). This is associated with conduction disease, atrial arrhythmias, heart failure and sudden death and should be suspected when DCM is accompanied by elevated serum creatine kinase, conduction disease or frequent arrhythmias. In these patients, evidence suggests an ICD should be considered at a much lower threshold than in other cases of DCM81-83. A multicentre cohort of 269 patients with LMNA mutation identified non-sustained VT, LVEF <45%, male gender and non-missense mutations as risk factors for malignant ventricular arrhythmias84. Some authorities suggest ICD implantation should be considered following even mild cardiac expression. A recent report suggests that titin (TTN) mutations are a common cause of DCM85. TTN mutations have long been considered candidate causes of cardiomyopathy, but the size of the gene and the presence of many allelic variants have made it difficult to study86-89. To overcome some of these difficulties, Herman and colleagues85 used next generation sequencing to analyse genomic TTN sequence for mutations that altered full length cDNA (truncating mutations) in 792 subjects (312 DCM, 231 HCM and 249 controls). Truncating TTN mutations were found more frequently in DCM (27%) than in HCM (1%) or controls (3%). TTN mutations cosegregated with DCM in families with penetrant disease, but were also found in 18% of apparently sporadic cases. TTN sequencing is therefore likely to have a prominent role in the genetic evaluation of DCM patients in order to facilitate prephenotypic diagnosis, but the high frequency in controls and sporadic cases suggests that the majority of TTN mutants may yet prove to be susceptibility rather than causative factors. The importance of epigenetic factors (ie, processes that alter gene activation without changing DNA sequence) has also been highlighted since 2011. In a study comparing genome-wide cardiac DNA methylation in patients with idiopathic DCM and controls, Haas et al detected differences in the methylation of genes implicated in heart failure pathways. These were associated with differences in mRNA expression, a finding further strengthened by studies in zebra fish90. Epigenetics research is particularly advanced in cancer biology, contributing to potential therapeutic/diagnostic biomarker and therapeutic targets91-93. Its potential in cardiovascular genetics is yet to be realised. Genetic predisposition to inflammatory myocardial damage is also emerging as an important theme94. The Coxsackie virus, a common cause of myocarditis, causes proteolysis of dystrophin in infected cardiomyocytes 95-97. Genetic defects of the dystrophin–glycoprotein complex, associated with muscular dystrophy, frequently cause a DCM where CMR findings are often indistinguishable from myocarditis 98. In another study, the presence of variants in Toll-like receptors which play a key role in the innate immune response were associated with poorer cardiac function in 158 patients99. Finally, Meder et al100 present data associating the locus containing major histocompatibility genes (MHC I and II) with DCM. The authors identified multiple single nucleotide polymorphisms on chromosome 6p21. A specific locus was identified and an association was found with closely located genes encoding class I and class II major histocompatibility complex heavy chain receptors.
Prediction of outcomes in DCM
As in other cardiomyopathies, the role of CMR in predicting outcomes is an active area of study in DCM. Recent evidence suggests that the presence of midwall LGE identifies a DCM cohort at increased mortality risk. During a median follow-up period of just over 5 years, 27% of 142 DCM with LGE patients died compared with 11% of 330 DCM patients without LGE101. The pathophysiological basis of this difference is unclear. Data describing the diagnostic and prognostic utility of CMR surrogate for other tissue abnormalities, such as oedema, diffuse fibrosis or myocyte disarray, are expected in the near future. Some of the most important data since 2011 have been in children with DCM. In a paediatric registry population of 1803 patients, a 5-year incidence rate of 29% for heart transplantation, 12.1% non-SCD and 2.4% for SCD are quoted. A risk stratification model for SCD based on echocardiography data had a sensitivity of 86% and 57% specificity. Significant factors were LV dilatation, age at diagnosis and posterior wall thinning102. Another study investigating 175 paediatric patients with DCM reports death or transplantation in 26% within 1 year of diagnosis with a survival free of death or transplant of 56% 20 years after diagnosis. An increased risk for death or transplant was associated with the age at diagnosis, presence of familial cardiomyopathy and lower baseline LV fractional shortening 103.
Advances in treatment
Stem cell therapy has been a major topic over the course of the last few years. A 5-year follow-up randomised controlled study of 110 patients with DCM shows improved LV function, improved exercise tolerance and greater long term survival in patients who underwent intracoronary stem cell transplantation. Total mortality was 14% in the stem cell group versus 35% in the controls with rates of pump failure of 5% versus 18%. There was no difference in rates of sudden death 104. A systematic review on 29 preclinical and 15 clinical studies scrutinised stem cell therapy as a treatment for DCM. The majority of studies showed a modest improvement of LVEF after cell therapy during follow-up. Due to the large heterogeneity in criteria for inclusion, procedure and outcome measure, the need for randomised controlled trials was stressed105. Effect of exercise has been evaluated in patients with DCM following an 8-week period of short term exercise. Significant improvement in cardiac function at rest and following exercise was reported with sedentary patients having the greatest improvement 106. The importance of immune activation is another focus for therapy in DCM. Atorvastatin in a small dose reduced the levels of inflammatory cytokines (IL-6, TNF α), uric acid and N-terminal pro-brain natriuretic peptide in a small cohort of patients with DCM107. Overall evidence suggests improving survival in patients with idiopathic DCM. A study of 603 patients over three decades gives evidence for the impact of clinical guidelines on morbidity and mortality. Patients were subdivided in four enrolment periods and a 42% risk reduction per enrolment interval with regard to heart failure-related mortality and sudden death was reported108.
SUMMARY
Cardiomyopathies continue to be an area of intense interest in the literature. While the spectrum of disease is considerable and continues to expand, the themes are very similar between cardiomyopathy subtypes, with great emphasis on accurate diagnosis, disease stratification and aetiology driven therapy. Advances in these areas will depend on discovery science and the application of omic technologies, but the greatest insights are likely to emerge from large scale multicentre collaborations. All the evidence suggests that the next few years will be one of considerable advance. Contributors: All the authors contributed to the following: conception and design, acquisition and inter pretation of data; drafting the article or revising it critically for important intellectual content; and final approval of the version to be published. Competing interests: OPG received research support from the British Heart Foundation. All the other authors have no conflicts of interest. Provenance and peer review: Commissioned; externally peer reviewed.
References
1. Maron BJ, Gardin JM, Flack JM, et al. Prevalence of hypertrophic cardiomyopathy in a general population of young adults: echocardiographic analysis of 4111 subjects in the CARDIA study. Circulation 1995;92:785–9.
2. Elliott P, McKenna WJ. Hypertrophic cardiomyopathy. Lancet 2004; 363:1881–91.
3. Guttmann OP, Rahman MS, O’Mahony C, et al. Atrial fi brillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart 2014;100:6 465–72.
4. Puntmann VO, Voigt T, Chen Z, et al. Native T1 mapping in differentiation of normal myocardium from diffuse disease in hypertrophic and dilated cardiomyopathy. JACC Cardiovasc Imaging 2013;6:475–84.
5. Sado DM, Flett AS, Banypersad SM, et al. Cardiovascular magnetic resonance measurement of myocardial extracellular volume in health and disease. Heart 2012;98:1436–41.
6. Sado DM, White SK, Piechnik SK, et al. Identification and assessment of Anderson-Fabry disease by cardiovascular magnetic resonance noncontrast myocardial T1 mapping. Circ Cardiovasc Imaging
2013;6:392–8.
7. Ferreira VM, Piechnik SK, Dall’armellina E, et al. Non-contrast T1-mapping detects acute myocardial edema with high diagnostic accuracy: a comparison to T2-weighted cardiovascular magnetic resonance. J Cardiovasc Magn Reson 2012;14:42.
8. Valente AM, Lakdawala NK, Powell AJ, et al. Comparison of echocardiographic and cardiac magnetic resonance imaging in hypertrophic cardiomyopathy sarcomere mutation carriers without left ventricular hypertrophy. Circ Cardiovasc Genet 2013;6:230–7.
9. Ho CY, Abbasi SA, Neilan TG, et al. T1 measurements identify extracellular volume expansion in hypertrophic cardiomyopathy sarcomere mutation carriers with and without left ventricular hypertrophy. Circ Cardiovasc Imaging 2013;6:415–22.
10. Greulich S, Schumm J, Grun S, et al. Incremental value of late gadolinium enhancement for management of patients with hypertrophic cardiomyopathy. Am J Cardiol 2012;110:1207–12.
11. Green JJ, Berger JS, Kramer CM, et al. Prognostic value of late gadolinium enhancement in clinical outcomes for hypertrophic cardiomyopathy. JACC Cardiovasc Imaging 2012;5:370–7.
12. van Veldhuisen DJ, Linssen GC, Jaarsma T, et al. B-type natriuretic peptide and prognosis in heart failure patients with preserved and reduced ejection fraction. J Am Coll Cardiol 2013;61:1498–506.
13. Geske JB, McKie PM, Ommen SR, et al. B-type natriuretic Peptide and survival in hypertrophic cardiomyopathy. J Am Coll Cardiol 2013;61:2456–60.
14. D’Amato R, Tomberli B, Castelli G, et al. Prognostic value of N-terminal pro-brain natriuretic peptide in outpatients with hypertrophic cardiomyopathy. Am J Cardiol 2013;112:1190–6.
15. Coats CJ, Gallagher MJ, Foley M, et al. Relation between serum Nterminal pro-brain natriuretic peptide and prognosis in patients with hypertrophic cardiomyopathy. Eur Heart J 2013;34:2529–37.
16. Kubo T, Kitaoka H, Yamanaka S, et al. Signifi cance of high-sensitivity cardiac troponin T in hypertrophic cardiomyopathy. J Am Coll Cardiol 2013;62:1252–9.
17. Nistri S, Olivotto I, Maron MS, et al. beta Blockers for prevention of exercise-induced left ventricular outflow tract obstruction in patients with hypertrophic cardiomyopathy. Am J Cardiol 2012;110:715–19.
18. Sherrid MV, Shetty A, Winson G, et al. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to fi rst-line therapy with beta-blockade or verapamil. Circ Heart Fail 2013;6:694–702.
19. Sorajja P, Ommen SR, Holmes DR Jr, et al. Survival aft er alcohol septal ablation for obstructive hypertrophic cardiomyopathy. Circulation 2012;126:2374–80.
20. Jensen MK, Prinz C, Horstkotte D, et al. Alcohol septal ablation in patients with hypertrophic obstructive cardiomyopathy: low incidence of sudden cardiac death and reduced risk profile. Heart 2013;99:1012–17.
21. Orme NM, Sorajja P, Dearani JA, et al. Comparison of surgical septal myectomy to medical therapy alone in patients with hypertrophic cardiomyopathy and syncope. Am J Cardiol 2013;111:388–92.
22. Iacovoni A, Spirito P, Simon C, et al. A contemporary European experience with surgical septal myectomy in hypertrophic cardiomyopathy.
Eur Heart J 2012;33:2080–7.
23. Desai MY, Bhonsale A, Smedira NG, et al. Predictors of long-term outcomes in symptomatic hypertrophic obstructive cardiomyopathypatients undergoing surgical relief of left ventricular outflow tract obstruction. Circulation 2013;128:209–16.
24. Galve E, Sambola A, Saldana G, et al. Late benefits of dual-chamber pacing in obstructive hypertrophic cardiomyopathy: a 10-year followup study. Heart 2010;96:352–6.
25. Mohiddin SA, Page SP. Long-term benefi ts of pacing in obstructive hypertrophic cardiomyopathy. Heart 2010;96:328–30.
26. Kappenberger LJ, Linde C, Jeanrenaud X, et al. Clinical progress after randomized on/off pacemaker treatment for hypertrophic obstructive cardiomyopathy. Pacing in Cardiomyopathy (PIC) Study Group. Europace 1999;1:77–84.
27. Maron BJ, Nishimura RA, McKenna WJ, et al. Assessment of permanent dual-chamber pacing as a treatment for drug-refractory symptomaticpatients with obstructive hypertrophic cardiomyopathy. A randomized, double-blind, crossover study (M-PATHY). Circulation 1999;99:2927–33.
28. Qintar M, Morad A, Alhawasli H, et al. Pacing for drug-refractory or drug-intolerant hypertrophic cardiomyopathy. Cochrane Database Syst Rev 2012; (5):CD008523.
29. Schinkel AF, Vriesendorp PA, Sijbrands EJ, et al. Outcome and complications after implantable cardioverter defi brillator therapy in hypertrophic cardiomyopathy: systematic review and meta-analysis. Circ Heart Fail 2012;5:552–9.
30. O’Mahony C, Lambiase PD, Quarta G, et al. The long-term survival and the risks and benefits of implantable cardioverter defibrillators in patients with hypertrophic cardiomyopathy. Heart 2012;98:116–25.
31. Bos JM, Maron BJ, Ackerman MJ, et al. Role of family history of sudden death in risk stratification and prevention of sudden death with implantable defibrillators in hypertrophic cardiomyopathy. Am J Cardiol 2010;106:1481–6.
32. Christiaans I, van Engelen K, van Langen IM, et al. Risk stratification for sudden cardiac death in hypertrophic cardiomyopathy: systematic review of clinical risk markers. Europace 2010;12:313–21.
33. O’Mahony C, Tome-Esteban M, Lambiase PD, et al. A validation study of the 2003 American College of Cardiology/European Society of Cardiology and 2011 American College of Cardiology Foundation/
American Heart Association risk stratifi cation and treatment algorithms for sudden cardiac death in patients with hypertrophic cardiomyopathy. Heart 2013;99:534–41.
34. Elliott PM, Gimeno JR, Tome MT, et al. Left ventricular outflow tract obstruction and sudden death risk in patients with hypertrophic cardiomyopathy. Eur Heart J 2006;27:1933–41.
35. Maron BJ, Rowin EJ, Casey SA, et al. Risk stratifi cation and outcome of patients with hypertrophic cardiomyopathy >=60 years of age. Circulation 2013;127:585–93.
36. Moak JP, Leifer ES, Tripodi D, et al. Long-term follow-up of children and adolescents diagnosed with hypertrophic cardiomyopathy: risk factors for adverse arrhythmic events. Pediatr Cardiol 2011;32:1096– 105.
37. Maron BJ, Spirito P, Ackerman MJ, et al. Prevention of sudden cardiac death with implantable cardioverter-defi brillators in children and adolescents with hypertrophic cardiomyopathy. J Am Coll Cardiol 2013;61:1527–35.
38. Ingles J, McGaughran J, Scuffh am PA, et al. A cost-effectiveness model of genetic testing for the evaluation of families with hypertrophic cardiomyopathy. Heart 2012;98:625–30.
39. Wordsworth S, Leal J, Blair E, et al. DNA testing for hypertrophic cardiomyopathy: a cost-effectiveness model. Eur Heart J 2010;31:926–35.
40. Jensen MK, Havndrup O, Christiansen M, et al. Penetrance of hypertrophic cardiomyopathy in children and adolescents: a 12-year follow-up study of clinical screening and predictive genetic testing. Circulation 2013;127:48–54.
41. Lopes LR, Rahman MS, Elliott PM. A systematic review and metaanalysisof genotype-phenotype associations in patients with hypertrophic cardiomyopathy caused by sarcomeric protein mutations.
Heart 2013;99:1800–11.
42. Liang P, Lan F, Lee AS, et al. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation 2013;127:1677–91.
43. Lan F, Lee AS, Liang P, et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient specific induced pluripotent stem cells. Cell Stem Cell 2013;12:101–13.
44. Corrado D, Basso C, Pilichou K, et al. Molecular biology and clinical management of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart 2011;97:530–9.
45. Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J 2010;31:806–14.
46. Protonotarios N, Anastasakis A, Antoniades L, et al. Arrhythmogenic right ventricular cardiomyopathy/dysplasia on the basis of the revised diagnostic criteria in aff ected families with desmosomal mutations. Eur Heart J 2011;32:1097–104.
47. Quarta G, Muir A, Pantazis A, et al. Familial e valuation in arrhythmogenic right ventricular cardiomyopathy: impact of genetics and revised task force criteria. Circulation 2011;123:2701–9.
48. Basso C, Corrado D, Th iene G. Arrhythmogenic right ventricular cardiomyopathy in athletes: diagnosis, management, and recommendations for sport activity. Cardiol Clin 2007;25:415–22, vi.
49. Zaidi A, Ghani S, Sheikh N, et al. Clinical significance of electrocardiographic right ventricular hypertrophy in athletes: comparison with arrhythmogenic right ventricular cardiomyopathy and pulmonary hypertension. Eur Heart J 2013;34:3649–56.
50. Munkho lm J, Christensen AH, Svendsen JH, et al. Usefulness of immunostaining for plakoglobin as a diagnostic marker of arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol 2012;109:272–5.
51. Asimaki A, Tandri H, Duff y ER, et al. Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm
Electrophysiol 2011;4:743–52.
52. Gomes J, Finlay M, Ahmed AK, et al. Electrophysiological abnormalities precede overt structural changes in arrhythmogenic right ventricular cardiomyopathy due to mutations in desmoplakin-A combined murine and human study. Eur Heart J 2012;33:1942–53.
53. Cox MG, van der Zwaag PA, van der Werf C, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: pathogenic desmosome mutations in index-patients predict outcome of family screening:
Dutch arrhythmogenic right ventricular dysplasia/cardiomyopathy genotype-phenotype follow-up study. Circulation 2011;123:2690–700.
54. Li Mura IE, Bauce B, Nava A, et al. Identifi cation of a PKP2 gene deletion in a family with arrhythmogenic right ventricular cardiomyopathy. iEur J Hum Genet 2013;21:1226–31.
55. van der Zwaag PA, van Rijsingen IA, de Ruiter R, et al. Recurrent and founder mutations in the Netherlands-Phospholamban p.Arg14del mutation causes arrhythmogenic cardiomyopathy. Neth Heart J
2013;21:286–93.
56. Baskin B, Skinner JR, Sanatani S, et al. TMEM43 mutations associated with arrhythmogenic right ventricular cardiomyopathy in non- Newfoundland populations. Hum Genet 2013;132:1245–52.
57. van Hengel J, Calore M, Bauce B, et al. Mutations in the area composita protein alphaT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur Heart J 2013;34:201–10.
58. Gerull B, Kirchner F, Chong J, et al. A homozygous founder mutation in desmocollin-2 (DSC2) causes arrhythmogenic cardiomyopathy in the Hutterite population. Circ Cardiovasc Genet 2013;6:327–36.
59. Haywood AF, Merner ND, Hodgkinson KA, et al. Recurrent missense mutations in TMEM43 (ARVD5) due to founder eff ects cause arrhythmogenic cardiomyopathies in the UK and Canada. Eur Heart J
2013;34:1002–11.
60. Groeneweg JA, van der Zwaag PA, Jongbloed JD, et al. Left -dominant arrhythmogenic cardiomyopathy in a large family: associated desmosomal or nondesmosomal genotype? Heart Rhythm 2013;10:548–59.
61. Quarta G, Syrris P, Ashworth M, et al. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur Heart J 2012; 33:1128–36.
62. Marcus FI, Edson S, Towbin JA. Genetics of arrhythmogenic right ventricular cardiomyopathy: a practical guide for physicians. J Am Coll Cardiol 2013;61:1945–8.
63. Kapplinger JD, Landstrom AP, Salisbury BA, et al. Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia-associated mutations from background genetic noise. J Am Coll Cardiol
2011;57:2317–27.
64. Kim C, Wong J, Wen J, et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 2013;494:105–10. 65. Corrado D, Basso C, Rizzoli G, et al. Does sports activity enhance the risk of sudden death in adolescents and young adults? J Am Coll Cardiol 2003;42:1959–63.
66. Fabritz L, Fortmuller L, Yu TY, et al. Can preload-reducing therapy prevent disease progression in arrhythmogenic right ventricular cardiomyopathy? Experimental evidence and concept for a clinical trial. Prog Biophys Mol Biol 2012;110:340–6.
67. Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004;109:2807– 16.
68. Maron BJ, Ackerman MJ, Nishimura RA, et al. Task Force 4: HCM and other cardiomyopathies, mitral valve prolapse, myocarditis, andMarfan syndrome. J Am Coll Cardiol 2005;45:1340–5.
69. James CA, Bhonsale A, Tichnell C, et al. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy associated desmosomal mutation carriers. J Am Coll Cardiol 2013;62:1290–7.
70. Fabritz L, Hoogendijk MG, Scicluna BP, et al. Load-reducing therapy prevents development of arrhythmogenic right ventricular cardiomyopathy in plakoglobin-defi cient mice. J Am Coll Cardiol
2011;57:740–50.
71. Dalal D, Jain R, Tandri H, et al. Long-term effi cacy of catheter ablation of ventricular tachycardia in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol 2007;50:432–40.
72. Verma A, Kilicaslan F, Schweikert RA, et al. Short- and long-term success of substrate-based mapping and ablation of ventricular tachycardia in arrhythmogenic right ventricular dysplasia. Circulation 2005;111:3209–16.
73. Philips B, Madhavan S, Ja mes C, et al. Outcomes of catheter ablation of ventricular tachycardia in arrhythmogenic right ventricular dysplasia/ cardiomyopathy. Circ Arrhythm Electrophysiol 2012;5:499– 505.
74. Basso C, Corrado D, Bauce B, et al. Arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol 2012;5:1233–46. 75. Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death). J Am Coll Cardiol 2006;48:e247–346.
76. Schinkel AF. Implantable cardioverter defi brillators in arrhythmogenic right ventricular dysplasia/cardiomyopathy: patient outcomes, incidence of appropriate and inappropriate interventions, and complications. Circ Arrhythm Electrophysiol 2013;6:562–8.
77. Te Riele AS, Bhonsale A, James CA, et al. Incremental value of cardiac magnetic resonance imaging in arrhythmic risk stratifi cation of arrhythmogenic right ventricular dysplasia/cardiomyopathy associated desmosomal mutation carriers. J Am Coll Cardiol 2013;62:1761–9.
78. Deac M, Alpendurada F, Fanaie F, et al. Prognostic value of cardiovascular magnetic resonance in patients with suspected arrhythmogenic right ventricular cardiomyopathy. Int J Cardiol 2013;168:3514–21.
79. Hong TT, Cogswell R, James CA, et al. Plasma BIN1 correlates with heart failure and predicts arrhythmia in patients with arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm 2012;9:961–7.
80. Bhonsale A, James CA, Tichnell C, et al. Risk stratifi cation in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. Circ Arrhythm Electrophysiol
2013;6:569–78.
81. van Berlo JH, de Voogt WG, van der Kooi AJ, et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med (Berl) 2005;83:79–83.
82. Meune C, Van Berlo JH, Anselme F, et al. Primary prevention of sudden death in patients with lamin A/C gene mutations. N Engl J Med 2006;354:209–10.
83. Taylor MR, Fain PR, Sinagra G, et al. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol 2003;41:771–80.
84. van Rijsingen IA, Arbustini E, Elliott PM, et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J Am Coll Cardiol 2012;59(5):493–500.
85. Herman DS, Lam L, Taylor MR, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med 2012;366:619–28.
86. Gerull B, Gramlich M, Atherton J, et al. Mutations of TTN, encoding the giant muscle fi lament titin, cause familial dilated cardiomyopathy. Nat Genet 2002;30:201–4.
87. Gerull B, Atherton J, Geupel A, et al. Identifi cation of a novel frameshift mutation in the giant muscle fi lament titin in a large Australian family with dilated cardiomyopathy. J Mol Med (Berl) 2006;84:478– 83.
88. Siu BL, Niimura H, Osborne JA, et al. Familial dilated cardiomyopathy locus maps to chromosome 2q31. Circulation 1999;99:1022–6.
89. Satoh M, Takahashi M, Sakamoto T, et al. Structural analysis of the titin gene in hypertrophic cardiomyopathy: identification of a novel disease gene. Biochem Biophys Res Commun 1999;262:411–17.
90. Haas J, Frese KS, Park YJ, et al. Alterations in cardiac DNA methylation in human dilated cardiomyopathy. EMBO Mol Med 2013;5:413–29.
91. Coppede F. Epigenetic biomarkers of colorectal cancer: focus on DNA methylation. Cancer Lett 2014;342:238–47.
92. Litzow MR. Novel therapeutic approaches for acute lymphoblastic leukemia. Hematol Oncol Clin North Am 2011;25:1303–17.
93. Herceg Z, Vaissiere T. Epigenetic mechanisms and cancer: an interface between the environment and the genome. Epigenetics 2011;6:804–19.
94. Mavrogeni S, Spargias C, Bratis C, et al. Myocarditis as a precipitating factor for heart failure: evaluation and 1-year follow-up using cardiovascular magnetic resonance and endomyocardial biopsy. Eur J Heart Fail 2011;13:830–7.
95. Badorff C, Knowlton KU. Dystrophin disruption in enterovirus-induced myocarditis and dilated cardiomyopathy: from bench to bedside. Med Microbiol Immunol 2004;193:121–6.
96. Badorff C, Fichtlscherer B, Rhoads RE, et al. Nitric oxide inhibits dystrophin proteolysis by coxsackieviral protease 2A through S-nitrosylation: a protective mechanism against enteroviral cardiomyopathy. Circulation 2000;102:2276–81.
97. Badorff C, Lee GH, Knowlton KU. Enteroviral cardiomyopathy: bad news for the dystrophin-glycoprotein complex. Herz 2000;25:227–32. 98. Mavrogeni S, Papavasiliou A, Spargias K, et al. Myocardial infl ammation in Duchenne Muscular Dystrophy as a precipitating factor for heart failure: a prospective study. BMC Neurol 2010;10:33.
99. Riad A, Meyer zu SH, Weitmann K, et al. Variants of Toll-like receptor 4 predict cardiac recovery in patients with dilated cardiomyopathy. J Biol Chem 2012;287:27236–43.
100. Meder B, Ruhle F, Weis T, et al. A genome-wide association study identifies 6p21 as novel risk locus for dilated cardiomyopathy. EurHeart J 2013.
101. Gulati A, Jabbour A, Ismail TF, et al. Association of fi brosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. JAMA 2013;309:896–908.
102. Pahl E, Sleeper LA, Canter CE, et al. Incidence of and risk factors for sudden cardiac death in children with dilated cardiomyopathy: a report from the Pediatric Cardiomyopathy Registry. J Am Coll Cardiol 2012;59:607–15.
103. Alexander PM, Daubeney PE, Nugent AW, et al. Long-term outcomes of dilated cardiomyopathy diagnosed during childhood: results from a national population-based study of childhood cardiomyopathy. Circulation 2013;128:2039–46.
104. Vrtovec B, Poglajen G, Lezaic L, et al. Effects of intracoronary CD34+ stem cell transplantation in nonischemic dilated cardiomyopathy patients: 5-year follow-up. Circ Res 2013;112:165–73.
105. Gho JM, Kummeling GJ, Koudstaal S, et al. Cell therapy, a novel remedy for dilated cardiomyopathy? A systematic review. J Card Fail 2013;19:494–502.
106. Holloway CJ, Dass S, Suttie JJ, et al. Exercise training in dilated cardiomyopathy improves rest and stress cardiac function without changes in cardiac high energy phosphate metabolism. Heart 2012;98:1083– 90.
107. Bielecka-Dabrowa A, Mikhailidis DP, Rizzo M, et al. Th e influence of atorvastatin on parameters of infl ammation left ventricular function, hospitalizations and mortality in patients with dilated cardiomyopathy— 5-year follow-up. Lipids Health Dis 2013;12:47.
108. Castelli G, Fornaro A, Ciaccheri M, et al. Improving survival rates of patients with idiopathic dilated cardiomyopathy in tuscany over three decades: impact of evidence-based management. Circ Heart Fail 2013; 6:913–21.
 This work is licensed under a
This work is licensed under a