Larisa Anghel1,2, Liviu Macovei1,2, Cristina Prisacariu1,2, Catalina Arsenescu Georgescu1,2
1 „Prof. Dr. George I. M. Georgescu” Institute of Cardiovascular Diseases, Iasi, Romania
2 „Grigore T. Popa” University of Medicine and Pharmacy Iasi, Romania
Abstract: Introduction – Familial hypercholesterolemia is an autosomal dominant disorder associated with a high prevalence of early coronary artery disease. Case report – We report a case of myocardial infarction in a young man with familial hypercholesterolemia and three severe coronary artery disease. The family history was negative for coronary artery disease but his father and sister were diagnosed with familial hypercholesterolemia. On admission, he was hemodyna-mically stable but his electrocardiograms revealed an acute anterior myocardial infarction with moderate left ventricular systolic disfunction on echocardiography. He was immediately transferred for coronary angiography which revealed three severe coronary lesions and we decided to perform primary coronary angioplasty of the culprit lesion. After three months of treatment with statin and ezetimibe, the lipid profile of the patient was significantly improved. Discussions – Due to the rarity of this condition and the severity of its complications, the present article aims to report a case of 34 years old male with chronic inferior myocardial infarction and accelerated atherosclerosis resulting in a new acute coronary event. Conclusion – Screening of first-degree relatives and extended family members plays an important role in early detection and treatment, in order to save the affected individual and the other family members from catastrophic cardiac events. Keywords: familial hypercholesterolemia, atherosclerosis, coronary disease, myocardial infarction
INTRODUCTION
Familial hypercholesterolemia (FH) is an autosomal dominant genetic disorder due to mutations in the LDL receptor gene located on chromosome 191, pre-sented by high levels of serum low density lipoprotein (LDL), xanthomas and early coronary artery disease (CAD). The main cause of FH is LDL receptor abnor-malities that decrease the uptake of LDL into cells, particularly into the liver cells, from the blood, resul-ting in the increase of serum LDL-cholesterol levels2,3. Atherosclerotic process has been observed to begin in early life and certain pediatric diseases are linked to accelerated atherosclerosis resulting in clini-cal coronary events. It has been observed that familial hypercholesterolemia is the most common primary hyperlipidemia linked to cardiovascular abnormalities in childhood, often in the first decade of life4. Coro-nary artery disease is a multifactorial disorder in which several intrinsic and extrinsic actors, such as age, sex, blood pressure, serum lipids, fat intake, cigarette smoking, and physical activity may modify the individual’s genetically determined risk. A number of population, case-control, and intervention studies have shown a positive correlation between serum low density lipo-protein cholesterol level and risk of CAD whereas there is an inverse relationship between serum high density lipoprotein (HDL) concentration and occur-rence of CAD. Therefore, the identifi cation and ma-nagement of FH in early age is of great importance4,5.
CASE PRESENTATION
We report the case of a 34-year-old male admitted to our institute complaining of retrosternal chest pain for three hours. The family history was negative for coro-nary artery disease but his father and his sister were diagnosed with familial hypercholesterolemia. Except for smoking (less than one pack of cigarettes per day) there was no reported history of cardiac risk factors. On admission, he was hemodynamically stable, with a blood pressure of 120/80 mmHg, heart rate 80 beats per minute, and oxygen saturation 96% on room air. Heart sounds were regular, with no murmur, click, bruit or rubs noted. The electrocardiograms revea-led the classic pattern of an acute anterior myocar-dial infarction, with an ST segment elevation of 5 mm in V1-V4 (Figure 1). A transthoracic echocardiogram demonstrated mild left ventricular systolic dysfunc-tion with an ejection fraction of 45%. Investigations revealed elevated cardiac enzymes and a severe lipid dysfunction with total cholesterol level of 331 mg/dL and low-density lipoprotein of 251 mg/dL. Complete hemogram, blood sugar, renal function test, liver func-tion test and thyroid function test were within normal limits. In this context, he was immediately transferred to the catheter laboratory for primary percutaneous coronary intervention (PCI). His coronary angiography revealed three severe coronary lesions: a thrombotic occlusion in the ostium of the left anterior descending coronary artery, a subocclusion of the postero-late-ral branch and chronic occlusion of the right artery (Figure 2). Under these circumstances we decided to perform PCI of the left anterior descending artery and balloon angioplasty to postero-lateral lesion, without periprocedural complications (Figure 3).
Initially, at the emergency room, the personal his-tory of the patient was noncontributory, but further history (after the coronary angiography) revealed that he had another chest pain four months ago, but wi-thout other medical controls and this may be caused by another acute coronary event and may explain the chronic occlusion of the right coronary artery.
After the myocardial revascularization, the patient was hemodynamically stable, without angina or ar-rhythmic events and was discharged home after five days. We began the chronic treatment with high dose of atorvastatin 80 mg/day, beta blocker, angiotensin converting enzyme inhibitor and double antiaplatelet agents.
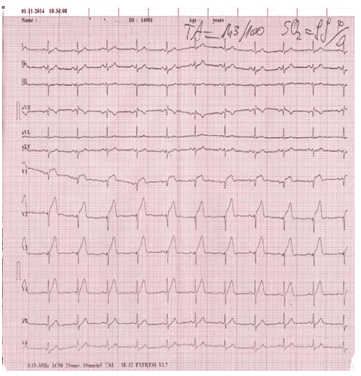
Figure 1. The admission ECG with the classic pattern of acute anterior myocardial infarction.
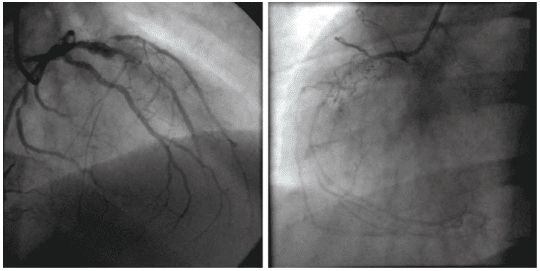
Figure 2. Coronary angiography: a thrombotic occlusion in the ostium of the left anterior descending coronary artery and subocclusion of the postero-lateral branch (A) and chronic occlusion of the right artery (B).
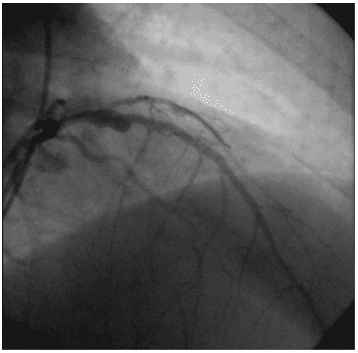
Figure 3. Coronary angiography: primary percutaneous coronary inter-vention of the left anterior descending artery and balloon angioplasty to the postero-lateral branch.
DISCUSSIONS
In the early 1970s, Goldstein et al. recognized that FH is a genetically determined defect within hepatocytes6. Specifically, he found that the LDL-receptor precur-sor was not transported to the cell surface in patients with FH; therefore, the LDL-receptors were incapable to bind LDL resulting in an extraordinary elevation in serum LDL6. There are two types of familial hyper-cholesterolemia: the heterozygous form in which the patient has one normal allele and one mutated allele is the most common form with an incidence of 1 out of 500, whereas the homozygous form in which the patient has two mutated alleles, considered an auto-somal codominant disorder, is rare with an incidence of approximately one in a million7. Patients with hete-rozygous FH are usually diagnosed as adults and often times respond well to medical therapy. On the other hand, patients with homozygous FH are often diagno-sed early in childhood, do not respond well to medical therapy, and can progress rapidly to premature coro-nary artery disease. The genetic mutations underlying FH affect the production and processing of cell surface LDL receptors resulting in impaired hepatic clearan-ce of circulating LDL particles, which leads to their accumulation in bloodstream. Elevated LDL levels are evident before birth and persist throughout the lifes-pan6,7. Furthermore, patients may develop accumulati-on of cholesterol in other parts of the body leading to the development of cutaneous xanthomas, which are most commonly located in the elbows, hands, knees, and Achilles tendon1.
Our patient had myocardial infarction at a young age of 34 years with a lipid profile comprising a high LDL-C and total cholesterol, with normal triglyceri-des. His family history was classical with 2 generations being affected and both male and female individuals be-ing affected equally. Hence this suggests an autosomal dominant disorder.
Simon Broome’s diagnostic criteria for familial hypercholesterolemia says a defi nite diagnosis of fa-milial hypercholesterolemia can be made if either the total cholesterol concentration is above 7.5 mmol/li-ter in adults or the LDL cholesterol concentration is above 4.9 mmol/liter in adults and if tendinous xantho-mas were present in the patient or a first degree rela-tive8-10. Our patient had total cholesterol levels of 8.56 mmol/l and a LDL-C of 6.50 mmol/l. His father and his elder sister had xanthomas. Severe LDLc elevations in our patient were in the absence of secondary causes of hypercholesterolemia, the triglyceride levels were within the reference range and HDL cholesterol levels were also within the reference range. Hence this con-firms the diagnosis of familial hypercholesterolemia in our patient. He should have had the heterozygous type of FH as this type presents much later in adult life with coronary artery disease and LDL-C levels are generally less than 400 mg/dl. Aggressive treatment of hypercholesterolemia including diet control, lipid lowering drugs, exercise and control of risk factors, will help to reduce the morbidity and mortality asso-ciated with this disease. Treatment options available for FH are lipid lowering drugs like statins, bile acid sequestrants, apheresis and liver transplantation. As liver is the most important tissue for removing circula-ting LDL, liver transplantation is an effective treatment option in this disorder9. Our patient was put initially on atorvastatin 80 mg daily. After six weeks follow up there was a regressi-on of the biochemical parameters (total cholesterol level from 331 mg/dL to 238 mg/dl and low-density lipoprotein of 251 mg/dL to 179 mg/dl). The current ESC Guidelines for the management of dyslipidemias11 recommend in patients with a very high cardiovascular risk an LDL-C goal less than 70 mg/dl or ≥50% LDL-C reduction when the target level cannot be reached. Also, the recommendations for patients with familial hypercholesterolemia are a high dose statin and whe-never needed in combination with cholesterol absorb-tion inhibitors and/or a bile acid sequestrant. Recently, some novel agents that target and inactivate proprote-in convertase subtilsin-kexin type 9 (PCSK9), a hepatic protease that attaches and internalizes LDL receptors into lysosomes hence promoting their destruction, have been approved. By preventing LDL receptor des-truction, LDL-C levels can be lowered 50%-60% abo-ve that achieved by statin therapy alone12,13. Hence in view of persistent LDL-C elevation inspite of maximal statin therapy for six weeks, we added the ezetimibe 10 mg/day, with a significant reduction of the LDL-C, from 331 mg/dl at admission to 119 mg/dl, 3 months after the acute coronary event.
Even it has been observed that familial hypercho-lesterolemia is the most common primary hyperlipi-demia linked to cardiovascular events in youth, the particularities of our case are the diagnostic of familial hypercholesterolemia in a context of acute coronary event, in a patient with a previous inferior myocardial infarction, diagnosed also in this context. In the same time, the patient has an accelerated atherosclerosis with three severe coronary lesions at a young age and also his son, a 3-years old boy, was screened and dia-gnosed with familial hypercholesterolemia.
Screening of first-degree relatives and extended family members plays an important role in early de-tection and treatment. Aggressive treatment of hyper-cholesterolemia including diet control, lipid lowering drugs, exercise and control of risk factors will help to reduce the morbidity and mortality associated with this disease11.
CONCLUSIONS
The major complication of familial hypercholesterole-mia is accelerated atherosclerosis, resulting in signifi-cant morbidity and mortality due to coronary artery diseases. Hence, early recognition is important and all the relatives in the family should be screened for dys-lipidemia. Early diagnosis and initiation of treatment will save the affected individual and the other family members from catastrophic cardiac events.
Conflict of interest: none declared.
Acknowledgements: The study is not supported by granting or commercial sources of funding.
Running title: The mystery of familial hypercholes-terolemia.
References
1. Nemati MH, Astaneh B. Optimal management of familial hypercho-lesterolemia: treatment and management strategies. Vasc Health Risk Manag 2010; 6(1):1079–1088.
2. Selvan JP, Uthaman B, Abushaban L, Jebaraj R. Homozygous familial hypercholesterolemia with generalized arterial disease. Med Princ Pract 2007; 16(1):75–78.
3. Khurana VK, Mehta RK, Chandra K. Homozygous familial hypercho-lesterolemia: Case report and Review of Literature. J Intern Med 2014; 2(2):34–40.
4. McMahan CA, Gidding SS, Fayad ZA, Zieske AW, Malcolm GT, Tra-cy RE. Risk scores predict atherosclerotic lesions in young people. Arch Intern Med 2005; 165(8):883–890.
5. Kraft HG, Lingenhel A, Raal FJ, Hohenegger M, Utermann G. Lipoprotein(a) in homozygous familial hypercholesterolemia. Arte-rioscler Thromb Vasc Biol 2000; 20(2):522–528.
6. Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver CR, Sly WS, Childs B, et al. The Metabolic and Molecular Basis of Inherited Disease. New York: McGraw-Hill, 2001: 2863– 2913.
7. DeMott K, Nherera L, Shaw EJ, Minhas R, Humphries SE, Kathoria M, Ritchie G, Nunes V, Davies D, Lee P, McDowell I, Neil A, Qureshi N, Rowlands P, Seed M, Stracey H, Thorogood M, Watson M. Clini-cal guidelines and evidence review for familial hypercholesterolae-mia: the identifi cation and management of adults and children with familial hypercholesterolaemia. 2008. London: National Collaborat-ing Centre for Primary Care and Royal College of General Practitio-ners.
8. Scientifi c steering Committee on behalf of the Simon Broome Regis-ter Group. Risk of fatal coronary heart disease in familial hypercho-lesterolemia. BMJ 1991; 303(6807):893–896.
9. Scientific Steering Committee on behalf of the Simon Broome Reg-ister Group. Mortality in treated heterozygous familial hypercho-lesterolemia: implications for clinical management. Atherosclerosis 1999; 142(1):105–112.
10. Shruthi B, Vimala SI, Girish I. Familial hypercholesterolemia – case report. Int J Med App Sci 2014; 3(1):180–184.
11. Catapano AL, Graham I, De Backer G. ESC/EAS Guidelines for the management of dyslipidaemias. The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Atherosclerosis 2016; 37(39):2999-3058.
12. Sbrana F, Dal Pino B, Bigazzi F, Ripoli A, Passino C, Gabutti A, Pas-anisi EM, Petersen C, Valleggi A, Todiere G, Barison A, Giannoni A, Panchetti L, Becherini F, Pianelli M, Luciani R, Sampietro T. Statin intolerance in heterozygous familial hypercolesterolemia with car-diovascular disease: After PCSK-9 antibodies what else? Eur J Prev Cardiol 2017; 24(14):1528-1531.
13. Chaudhary R, Garg J, Shah N, Sumner A. PCSK9 inhibitors: a new era of lipid lowering therapy. World J Cardiol 2017; 9(2):76-91.
 This work is licensed under a
This work is licensed under a