Alin Alexandru Ionescu1, Monica Chivulescu1, Alina Nicula2,3, Bogdan Alexandru Popescu1,3, Carmen Ginghina1,3, Ruxandra Jurcut1,3
1 “Prof. Dr. C.C. Iliescu” Emergency Institute for Cardiovascular Diseases, Bucharest, Romania
2 Department of Radiology, Emergency University Hospital, Bucharest, Romania
3 “Carol Davila” University of Medicine and Pharmacy, Bucharest, Romania
Abstract: Arrhythmogenic right ventricular dysplasia (ARVD), with the more appropriate label of ‘arrhythmogenic cardiomyopathy’ (AC), is still considered a rare pathological entity. It is an inheritable heart muscle disease with a powerful negative impact on the survival and the quality of life of patients, primarily through malignant arrhythmias or, more rarely, the development of heart failure. We present the case of a man who was diagnosed with AC at the age of 32 years following a detailed cardiac evaluation prompted by an incidental fi nding of anabnormal ECG tracing (negative T waves from V1 through V3). Both the echocardiography and the cardiac MRI performed at that time revealed predominantly the involvement of the LV, with none or minor structural and functional RV defects. After 4 years, the disease progressed and now we are facing biventricular involvement, with moderate systolic dysfunction of the LV and RV, yet the patient remains asymptomatic. A series of steps including multimodality imaging techniques, various tests, family history and other aspects are needed in order to correctly stratify the risk of sudden cardiac death (SCD) which will ultimately translate into an appropriate therapeutic choice.
Keywords: arrhythmogenic right ventricular dysplasia (ARVD), arrhythmogenic cardiomyopathy (AC), biventricular dysfunction, cardiac MRI, arrhythmic risk stratifi cation.
INTRODUCTION
A growing body of knowledge points to some of the shortcomings of the revised Task Force Criteria 1 for diagnosing RVAD from 2010, the most important of which being the increasing need to recognize phenotypes with biventricular involvement and ever more so those with predominantly LV dysfunction, to take their particular aspects into account when considering the diagnosis and to aptly evaluate these patients in terms of arrhythmic risk.
CASE REPORT
We report the case of a 37-year-old male patient, for whom the suspicion of AC was raised 4 years earlier after a complete cardiac exam performed in response to subtle changes in the ECG tracing, which was part of a routine evaluation before minor surgery (laparoscopic cholecystectomy). At that time, the ECG showed negative T waves in V1 through V3, minor LBBB (with a QRS duration
of 100ms) and a normal QRS axis of 45 degrees (Figure 1). The patient had multiple cardiovascular risk factors, such as smoking, obesity and dyslipidemia, but had no significant cardiovascular diseases or any other comorbidities. Even though the threshold for a family history of SCD is <35 years, it is worth mentioning that the patient’s mother and father died at 53 and 46, without any clear cause, but apparently not suddenly. Nevertheless, the echocardiography revealed mild LV systolic dysfunction, with a slightly reduced LVEF of 50%, a thin myocardial wall at the level of the LV apex with dyskinesia in the form of an apical microaneurysm, but with normal diastolic function and chamber dimensions. Only a slight decrease of the RV systolic function was described.
All of these fi ndings were confi rmed and complemented by cardiac magnetic resonance (CMR) imaging, which described an LV with normal global systolic function but limitedapical dyskinesia and hypokinesia at the mid and apical segments of the lateral wall of the LV and the apical segment of the interventricular septum. T1 phase-sensitive inversion-recovery (PSIR) sequences showed late gadolinium enhancement (LGE), a marker for fibrosis, distributed at subendocardial level of mid segment of the infero-lateral wall, as well as midwall at the mid segment of the lateral wall. Consistent with the echocardiographic findings, there were no wall motion abnormalities (WMA), nor any signs of fi brous scars in the RV.
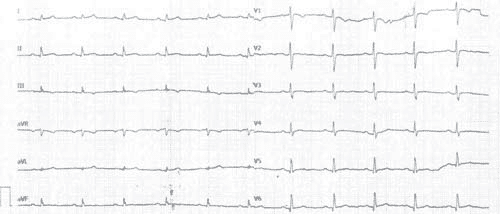
Figure 1. ECG tracing from 2012 – negative T waves in V1 through V3, minor LBBB (with a QRS duration of 100 ms) and a normal QRS axis.
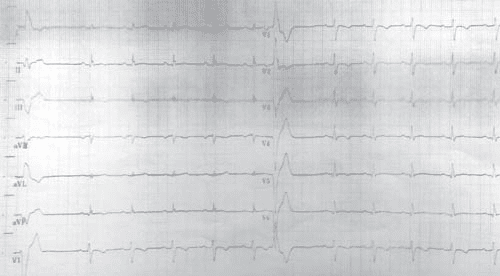
Figure 2. ECG tracing from 2016 – displays negative T waves from V1 through V6 and inferior leads (DII, DIII, aVF), with a PVC with RBBB appearance, with a fragmented appearance of the QRS complex and low voltage QRS complex on peripheral leads.
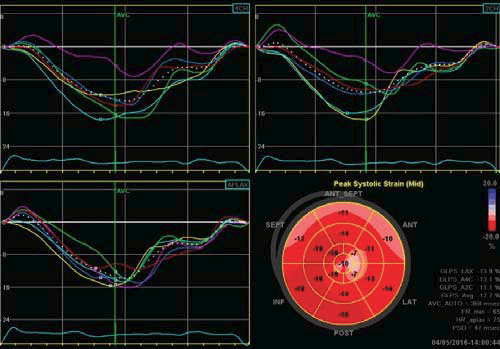
Figure 3. Strain rate imaging of the left ventricle with a bull’s eye map which shows an impaired cardiac deformation, with anaverage LV GLS = -12.7%.
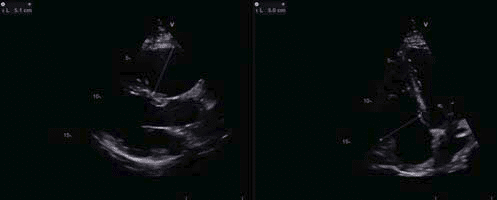
Figure 4. Transthoracic echocardiography showing an enlarged right ventricle, with large diameters both in parasternal long axis view and in apical four chambers view
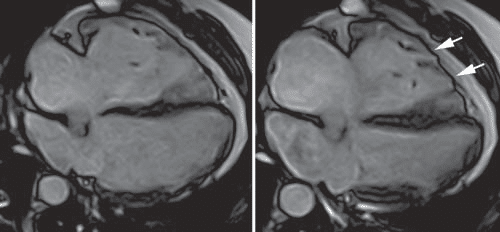
Figure 5. Cardiac magnetic resonance, four chamber view in diastole (A) and systole (B), displaying an enlarged right ventricle, several microaneurysms at the mid and apical segments of the RV free wall (arrows).
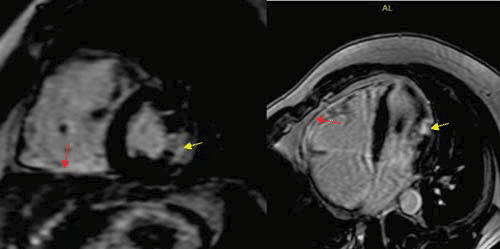
Figure 6. Cardiac magnetic resonance, LGE (BH 3D LGE 1.9 SA sequence, 4CH 2D Delayed), displaying mild delayed gadolinium enhancement at the level of the inferior and free RV wall (consistent with fibrosis – red arrow) and a transmural gadolinium enhancement at the level of the mid segment of thelateral LV wall (consistent with a small scar of myocardium infarction
– yellow arrow).
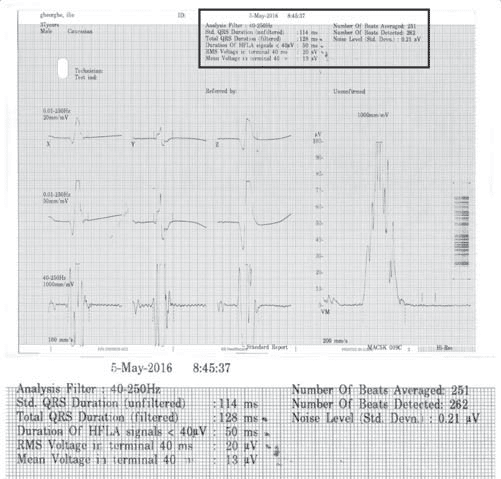
Figure 7. Signal-averaged ECG – the patient met 3/3 criteria.
After this extensive evaluation, the patient was lost to follow-up for almost 4 years, and came back for a routine evaluation in 2016. At present, the investigations included in the usual work-up of patients with ARVD (AC) suspicion were performed as recommended in the International Task Force consensus statements1,2. At this point, clear signs of disease progression became apparent. The ECG displayed negative T waves from V1 through V6 and the inferior leads (DII, DIII, aVF), with a PVC with RBBB appearance, with a fragmented appearance of the QRS complex and low voltage QRS complex on peripheral leads (Figure 2). The echocardiography revealed, apart from what was previously known, a pseudonormal fi lling pattern of the LV and a pathological average LVGLS of -12,7% (Figure 3). There was a more prominent involvement of the RV, which was enlarged (Figure 4) with global systolic dysfunction refl ected by a FAC of 22% and with accompanying subtle WMA, mostly apical. The CMR examination confi rmed these fi ndings, exhibiting an enlarged RV (RVEDVI of 130 ml/m2) with a pronounced systolic dysfunction (RVEF of 13%), with several microaneurysms at the mid-segments of the RV free wall and fi ne bands of fi brosis (Figure 4). There was a signifi cant progression of the disease concerning the LV, which displayed an EF of 37.5%, with multiple WMA, small apical aneurysm and localized fi brosis of the lateral wall. The 24-hour ECG monitoring,showedventricular premature contractions (750/24h), less than 4 years ago – most likely in response to optimization of therapy with beta-blockers. The ECG stress-test was unremarkable, with good exercise tolerance and no arrhythmic events. The signal-averaged ECG (SAECG), which is a practical and widely available tool to helpAC diagnosis, was performed and our patient met 3 out of 3 criteria (Figure 5), only 1 out of 3 being necessary to count as a minor criteria. At this point, according to the revised 2010 TFC1 the diagnosis is unequivocal, with the fulfi llment of 2 major imaging criteria (enlarged RV with suggestive WMA described on both echocardiography and CMR), 1 major ECG criteria (with inverted T waves in multiple leads) and several minor criteria, such as positive SAECG and over 500 PVCs on 24 hour rhythm monitoring. Nevertheless, the patient is completely asymptomatic, as he denies breathlessness, palpitations, syncope or chest pain. The patient was continued on an optimized beta-blocker dose (metoprolol succinate, 50 mg once daily). Seeing as he has several minor risk factors (proband, male gender, inverted T-waves in more than 3 precordial leads, fragmented QRS on ECG) and one major risk factor represented by moderate biventricular systolic dysfunction, the patient has a class IIafor ICD implantation based on expert consensus. Based on the 2015 ESC Guidelines on sudden death prevention3, in the absence of ventricular tachycardia, risk factors among which extensive RV disease, LV dysfunction and late gadolinium enhancement on CMR (including
LV involvement) only give a class IIb indication for ICD implantation.
DISCUSSIONS
Arrhythmogenic cardiomyopathy has no pathognomonic sign or gold-standard evaluation, and is thus diagnosed by gathering a series of elements that were included in the revised 2010 Task Force Criteria1. This paper recognizes ARVD as the most prominent variant of an otherwise complex and variable disease, but it doesn’t succeed in offering conclusive diagnostic criteria for the non-classical patterns, nor does it incorporate contrast-enhanced CMR4. There are increasingly expanding insights that already prove useful in identifying such phenotypes. From a clinical standpoint, LV involvement usually carries a higher risk of signifi cant arrhythmic events, which could be correlated with the degree of systolic dysfunction. Highly suggestive is the variable association of ECG characteristics, such as inverted T waves in the lateral or inferolateral leads (V5, V6, DI, aVL), low voltage QRS complex in the peripheral leads and PVCs with RBBB appearance5. While echocardiography could play a valuable role, CMR appears as the most sensitive and specifi c tool6. If using contrast-enhanced sequences, it can even pick up early signs of the disease, most often with a regional pattern at the level of the inferolateral or inferoseptal regions of the LV, with a propensity for subepicardial and midwall layers7. Because of the low prevalence of the disease, practical limitations hinder the development of RCT on this population with the consequence that most of the recommendations concerning treatment are based on data derived from case series, registries and heavily rely on the opinion of experts in the field. An International Task Force consensus paper regarding treatment for patients with AC was published in 2015 and deals with very important issues and outline the main therapeutic resources, including lifestyle modifi cation, betablockers and antiarrhythmic drugs (AAD), heart failure therapy, ICD, ablation and cardiac transplant. Patients with a defi nite diagnosis of AC should be restricted from participating in competitive sports8 and should refrain from performing endurance training, all the while maintaining the option of undergoing low intensity recreational sports. Beta-blockers represent first-line therapy as they are recommended for all those diagnosed with AC (IIa), but not for asymptomatic gene carriers; they also aid in case of ICD shocks, both appropriate and inappropriate, due to tachyarrhythmia. Even though there are some reports that sotalol is associated with better results and less long-term side- effects, the majority of the available evidence still supports amiodarone as the most effi cient drug of use9, and it should be administered, alone or in combination with beta-blockers, in order to improve symptoms in the case of frequent PVCs or as an adjunct therapy to ICD with frequent appropriate therapies. Nevertheless, pharmacological therapy does not represent an adequate substitute for ICD in those patients who warrant such an intervention10. Taking into account the short and long-term complications of ICDs and the fact that they would more often be employed in a relatively young segment of the population, clear recommendations have been prepared by expert consensus in order to aid with risk stratifi cation and proper selection of those who would benefi t the most after such therapy, usually a single chamber device. Those who have experienced an aborted SCD due to VF, episodes of sustained VT or who associate severe dysfunction of RV, LV or both require prophylaxis of SCD with an ICD because they have a risk of >10%/year to develop a major arrhythmic event. Those who have no
risk factors and those who are healthy gene carriers have the lowest arrhythmic risk of <1%/year and do not require an ICD. Between these 2 categories lies a gray area that can be further divided into two distinct situations. ICD should be taken into consideration (IIa) for patients who present with at least one major risk factor, represented by unexplained syncope, documented non-sustained VT, moderate systolic dysfunction of either/both ventricles. The presence of more than one minor risk factor could lead to the use of an ICD (IIb) and this takes into consideration male gender, a complex genotype (compound or digenicheterozygosisity), proband status, inducible VT/VF at programmed ventricular stimulation, extent of electroanatomic scar or fragmented electrograms on RV endocardial voltage mapping, extent of T-wave inversion (more than V1-V3) and T-wave inversion in inferior leads, QRS fragmentation and others. In our case, the patient has a IIa indication for primary prophylaxis of SCD with an ICD. Cardiac transplantation11 in this population is a somewhat rare occurrence and its main indications are progression of the disease to end-stage heart failure (almost 2/3 of patients that require heart transplant) or intractable ventricular arrhythmias that cannot be suppressed by the use of previously mentioned therapies.
A central aspect of any rare genetic cardiovascular disease is represented by family screening12,13, seeing as patients who are diagnosed in the early stages of the disease derive the most benefi t from a timely implementation of the therapeutic protocols mentioned above. If a mutation associated with AC is identifi ed, then cascade genetic testing of fi rst degree relatives is indicated.
If no such mutation is found or if the results are equivocal (which is true in almost half cases even in experienced centers), cascade cardiac evaluation of fi rst degree relatives is recommended periodically at specifi c intervals, taking into account several factors, such as age, degree of physical activity, symptoms and others.
CONCLUSIONS AND IMPLICATIONS FOR CLINICAL PRACTICE
Arrhythmogenic cardiomyopathy is a challenging disease, both from a diagnostic and therapeutic point of view, as it can require an elaborate array of tests to confi rm its presence and it entails the use of a wide range of treatment options. Further refi nements of the revised TFC from 2010 would be benefi cial, as they don’t provide an adequate recognition of AC phenotypes that involve predominantly the LV. Therapeutic options include the limitation of sports participation, the use of beta-blockers and AAD, ICDs for high risk patients, catheter ablation for frequent appropriate ICD shocks and heart transplant for endstage heart failure or intractable arrhythmias. We emphasize the role of family screening as a cornerstone in the management of patients with genetic
diseases. Taking all of this into account, we encourage that they should be evaluated and provided treatment preferably in expert centers that are frequently engaged with the particular diffi culties posed by these pathologic entities.
Acknowledgements: None declared.
Conflict of interests: none declared.
References
1. Marcus FI, McKenna WJ, Sherrill D et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modifi cation of the Task Force Criteria. Eur Heart J. 2010;31:806–14.
2. Corrado D, Wichter T, Link MS et al. Treatment of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: An International Task Force Consensus. Statement.Circulation. 2015;132:441–53.
3. Silvia G. Priori, Carina Blomström-Lundqvist et al. 015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC)Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015 1;36(41):2793-867
4. Pilichou K, Thiene G, Bauce B et al. Arrhythmogenic cardiomyopathy. Orphanet J Rare Dis. 2016 Apr 2;11:33.
5. Sen-Chowdhry S, Syrris P, Prasad SK, et al. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol. 2008;52:2175–87.
6. Perazzolo Marra M, Rizzo S, Bauce B et al. Arrhythmogenic right ventricular cardiomyopathy: Contribution of cardiac magnetic resonance imaging to the diagnosis. Herz. 2015;40:600–6.
7. PerazzoloMarra M, Leoni L, Bauce B et al. Imaging study of ventricular scar in arrhythmogenic right ventricular cardiomyopathy: comparison ofn3D standard electroanatomical voltage mapping and contrastenhanced cardiac magnetic resonance. CircArrhythm Electrophysiol. 2012;5:91–100.
8. Corrado D, Basso C, Rizzoli G et al. Does sports activity enhance the risk of sudden death in adolescents and young adults? J Am CollCardiol 2003;42: 1959–1963.
9. MarcusGM, Glidden DV, PolonskyB, et al. Efficacy of antiarrhythmic drugs in arrhythmogenic right ventricular cardiomyopathy: a report from the North American ARVC Registry.J Am CollCardiol 2009;54: 609–615
10. Corrado D, Leoni L, Link MS et al. Implantable cardioverter-defi brillator therapy for prevention of sudden death in patients with arrhyth mogenic right ventricular cardiomyopathy/dysplasia. Circulation 2003; 108:3084–3091
11. Tedford RJ, James C, Judge DP et al. Cardiac transplantation in arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am CollCardiol2012; 59:289–290.
12. Marcus F, Mestroni L. Family members of patients with ARVC: who is at risk? At what age? When and how often should we evaluate to determine risk? J Am CollCardiol 2014; 64:302.
13. Haugaa KH, Bundgaard H, Edvardsen Tet al. Management of patients with Arrhythmogenic Right Ventricular Cardiomyopathy in the Nordic countries, Scandinavian Cardiovascular Journal, 49:6, 299-307.
 This work is licensed under a
This work is licensed under a