Elena Popa1,2, Ana-Maria Daraban2,3, Roxana Enache1, Sorin Giuşcă1, Carmen Ginghină1,2, Ruxandra Jurcuţ1,2
1 Cardiology Department, Emergency Institute for Cardiovascular Diseases “Prof. Dr. C. C. Iliescu”
2 University of Medicine and Pharmacy “Carol Davila”, Bucharest, Romania
3 Internal Medicine Department, Clinical Emergency Hospital Bucharest, Romania
INTRODUCTION
In pulmonary hypertension (PH), right ventricular (RV) function is correlated with functional status and is a strong predictor of survival, whereas the pulmonary artery (PA) pressures are not strongly correlated with symptoms or outcome. Even if treatment decreases pulmonary arterial resistance, this is not necessarily translated in improvement of outcome1. On the other hand, pressure and resistance represent only one aspect of ventricular afterload, the other one being arterial compliance, as reflected by stiffness parameters. Therefore, it is important to study the RV and the pulmonary arterial tree as a unit, with all their aspects, to further the understanding of the physiopathology of PH and to eventually improve the outcome of patients with PH.
A. General aspects of right ventricular afterload
Ventricular afterload can be evaluated by several methods, for instance by the maximum ventricular wall stress or by the assessment of arterial hydraulic load. Wall stress is approximated using a transposition of the law of Laplace as follows: the maximum value of the product of volume and pressure divided by wall thickness. This transposition is problematic for the RV because it stems from Laplace’s law for spheric structures, while the RV has considerable regional variations in internal radius2. Assessment of pulmonary arterial hydraulic load depends on the particular aspects of the pulmonary circulation. The major anatomical differences between the systemic and pulmonary circulation are represented by the intrapulmonary location of the majority of pulmonary vessels and the extensive branching of the pulmonary vasculature. While the proximal great arteries share an intrathoracic course, thus being both subject to intrathoracic pressure variations, the pulmonary arterial tree is also subject to gravitational and radial distribution of interstitial and perivascular pressure. Moreover, the pulmonary arterial tree consists of a relatively short elastic proximal PA, with a complex pattern of vascular division, with important consequences on pulse wave transmission and wave reflection3. Functionally, ventricular afterload has two components: the “steady-fl ow” component, which represents the opposition that the ventricle faces in maintaining a forward flow with the observed mean flow rate, and the “pulsatile” component, which is added by the pulsatile nature of flow and dependent on the compliance of the arterial system and on wave reflections. While the first component is quantified as vascular resistance and is a part of clinical practice, the pulsatile afterload is often ignored, since it requires complex investigations of stiffness parameters. Nevertheless, the pulsatile afterload is especially important in the pulmonary circulation, given its low resistance-high compliance nature, as is emphasized by the pulsatile ratio of hydraulic power used by the ventricles to maintain flow: in the systemic circulation, approximately 10% of the hydraulic power generated by the left ventricle (LV) is used for the pulsatile component of flow4, whereas in the pulmonary circulation, the RV spends approximately 25%-30% of its power for the same purpose5,6. On the other hand, pulsatile power fraction increases in systemic hypertension7,8, while it remains constant in PH6.
The amount of hydraulic power spent to overcome the steady component is consumed at the level of arterioles and capillaries for both the systemic and pulmonary circulation, while the pulsatile component depends on the significantly different distribution of compliance in the two arterial systems. As mentioned before, extensive branching of the pulmonary circulation leads to a larger number of peripheral vessels, up to 8-10 times more in the pulmonary than in the systemic arterial tree. In the systemic arterial tree, 80% of total compliance is based in the thoracic-abdominal aorta9, while in the pulmonary circulation at least half of pulmonary arterial compliance is distal to the proximal large arteries9,10. Thus, the pulsatile component of ventricular hydraulic power is spent mostly at the level of the aorta in the systemic circulation and along the entire pulmonary arterial tree in the lesser circulation. This anatomical difference be tween the 2 major arterial systems leads to a constant product of resistance and compliance (, the arterial time-constant, expressed in seconds) in the pulmonary circulation, parameter that characterizes the decay of pressure in diastole. Graphically, this means that resistance (R) and compliance (C) are inversely related by a hyperbola, because of the inverse exponential relationship between the 2 parameters (Fig. 1). The RC-time remains constant in healthy individuals, in patients with PH and even after treatment of PH11,12. This means that in the early stage of PH, a small increase in pulmonary vascular resistance (PVR) leads to a large decrease in compliance, whereas in later stages, larger increases in PVR lead to only modest decreases in compliance, probably because at a certain level of PA pressure and dilation the PA approaches its elastic limit. From a clinical perspective, this implies that in early stages of PH the decrease in compliance may be more easily detected than the rise in PVR; conversely, in early stages, the same reduction in resistance leads to more significant increases in compliance than in later stages of PH. On the other hand, the level of PAP after which rises in pressure lead to insignificant decreases in compliance are approximately 40 mmHg for mean PAP or 60 mmHg for systolic PAP13,14. Another consequence of the constant RC-time in pulmonary circulation is the proportionality between systolic, mean and diastolic PAP in healthy individuals and in PH, while in the systemic circulation, where the RC-product varies with aortic pressure, there is no proportional relationship of systemic arterial pressures. On the contrary, in old age hypertension, systemic systolic pressure rise is frequently accompanied by a decrease in diastolic pressure, with a significant increase in pulse pressure (PP). On the other hand, the PP in the aorta is approximately 40% of mean pressure, while in the pulmonary circulation, the PP is approximately equal to mean pressure. Another difference between the systemic and the pulmonary circulation is the much larger range of pressure and compliance changes in the pulmonary arterial tree than in the systemic circulation. In systemic hypertension, resistance increases by one-fifth and compliance decreases 3-fold, while in PH, resistance and compliance can increase, respectively decrease approximately 20-times9. Thus, the load imposed on the RV in PH is proportionally much larger and the adaptation of the RV to increased afterload is more difficult than for the LV.
B. Evaluation of right ventricular pulsatile afterload
B.1. Pulmonary artery dilation in pulmonary hypertension
Pulmonary artery dilation is one of the consequences of PH and has been used as a non-invasive sign of PH. Good correlation has been shown between CT measurements of the main pulmonary vessels and the level of mean PAP; the dimensions of main PA and its main branches have been proposed for diagnosing PH and even for estimating the level of pressure overload15,16.
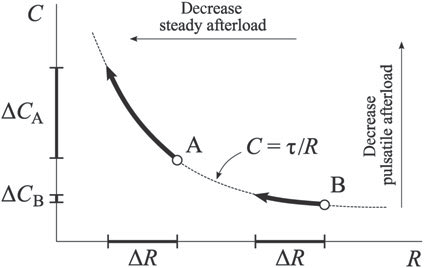
Figure 1. The inverse exponential relationship between C (compliance) and R (resistance). The same decrease in resistance (ΔR) leads to different decreases in compliance (ΔC), depending on the absolute levels of resistance. For high resistance values, the amplitude of compliance decrease is much smaller than for low resistance values. (reproduced with permission from 12: Lankhaar JW, Westerhof N, Faes TJ, Gan CT, Marques KM, Boonstra A, et al. Pulmonary vascular resistance and compliance stay inversely related during treatment of pulmonary hypertension. Eur Heart J 2008;29(13):1688-95.).
Later similar findings were reported using MR imaging17,18. The most used parameter is the ratio between the diameter of main PA and the diameter of ascending aorta; if over 1, the sensitivity and specificity of identifying mean PAP over 20 mmHg are 70% and 92% respectively, independently of body surface area and sex19. Although PAP is considered the main cause of PA dilation, the infl uence of other factors, such as time and flow, is less known. A recent study has shown that in patients treated for PH, the PA has continued to dilate, irrespective of changes in mean PAP, even in patients whose PAP has decreased with treatment20. Thus, PA dimensions are useful for screening for PH, but cannot be used for assessing the evolution of the disease or the therapeutic response. The causes for this behaviour of PA dilation during the course of PH are not completely understood. Histologic studies have shown significant remodeling of the proximal PA wall in PH, with extracellular matrix changes in collagen and elastin content, with significant increases in PA stiffness21,22. These structural changes might eventually become a cause of continued PA dilation, independent of changes in pressure and flow.
B. 2. Pulmonary artery input impedance
The ventricular afterload can be represented by the relationship between arterial pressure and flow, which can be expressed in the frequency or in the time domain. The most complete characterization is represented by the input impedance spectrum, a frequency domain analysis of the arterial circulation, which describes the relationship between arterial pressure and flow over an entire cardiac cycle, including both mean and pulsatile data. Initially, pulmonary impedance spectra were measured only in research laboratories using main PA catheterization, by simultaneously evaluating pulmonary arterial pressure and fl ow using high-fi delity, manometertipped catheters and fl owmeters. More recently, another approach has been validated, using instantaneous PAP measurements during right heart catheterization (RHC), combined with PA blood flow measurements using Doppler transthoracic echocardiography.
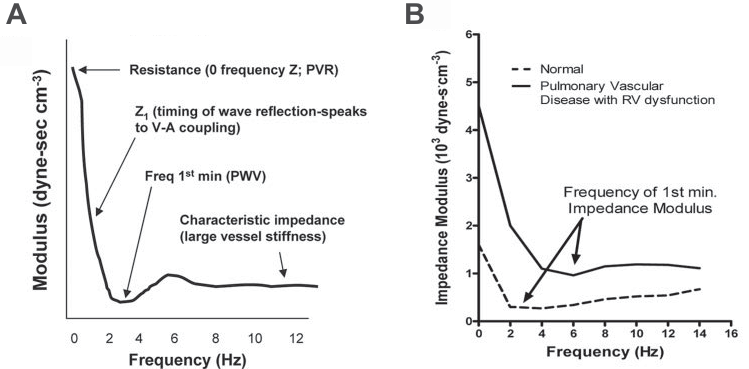
Figure 2. A. PA impedance spectrum in a normal subject. B. PA impedance spectra of a patient with PH and RV dysfunction (solid line) and normal subject (dashed line). Of note, the spectrum of the PH patient is shifted to higher than normal pressures and frequencies. (reproduced with permission from 26: Champion HC, Michelakis ED, Hassoun PM. Comprehensive invasive and noninvasive approach to the right ventricle-pulmonary circulation unit: state of the art and clinical and research implications. Circulation 2009;120(11):992-1007).
Consequently, Fourier analysis of pressure and flow data and computation of impedance are made using dedicated software23,24. The results of this analysis are expressed as an impedance spectrum, consisting of a pressure to flow ratio and a phase angle, expressed as a function of frequency25. The PA input impedance spectrum depends mainly on the fi rst 5 orders of bifurcation from the main PA and includes: a measure of total PVR (Z0 – 0 frequency impedance); indices of wave reflection (the first minimum of the ratio of pressure and flow or low-frequency phase angle, which also correspond to optimal ventricular-arterial coupling); characteristic impedance (Zc), which corresponds to the ratio of inertance to compliance; hydraulic load, as assessed by low-frequency impedance and the amplitude of impedance oscillations26 (Fig. 2). In patients with PH, the impedance spectrum in shifted to higher than normal pressures and frequencies, with higher Z0, reflecting higher PVR; a shift to higher frequencies of the point in which impedance declines to its fi rst minimum, reflecting increased pulse wave velocity and increased wave reflection; higher Zc, a measure of higher characteristic impedance; signs of increased mean and total hydraulic power output (3). In clinical practice, pulmonary impedance data have been shown in a pediatric population to better predict outcome in PH than PVR alone24. Although impedance spectra offer a wealth of data regarding arterial load, they are complex to interpret. Thus, other measures of pulsatile afterload have been defined, that can be more easily used in clinical practice.
B. 3. Time-domain analysis of pulmonary artery pressure waveforms
Time-domain analysis of pulmonary arterial pressure waveform provides important data on pulsatile arterial load and may be a surrogate for the full assessment of RV input impedance. This analysis can be performed using RHC. Pressure waveform analysis reveals the timing and extent of wave refl ection in the arterial circulation and the magnitude of pulse pressure (PP)27. Pulmonary artery pulse wave velocity (PWV) is around 1.7 m/s at normal pressures5, which is almost half of the velocity measured in the aorta at around 4 m/s28,29. The reflected pressure wave arrives back in both the aorta and the PA of normal subjects during the early diastolic period, because the shorter path length in the pulmonary arterial tree compensates for the lower pulse wave velocity27,30. When the forward pressure wave from the ventricle collides with the reflected backward pressure wave from the bifurcations, pressure increases and flow decreases26. Pressure and flow curves are more similar in the normal pulmonary arterial tree than in the systemic circulation, because of less wave reflection.
When reflection increases, because of pharmacological or pathological increases in arterial resistance, the resemblance of pressure and fl ow waves decreases30. The characteristics of the reflected wave depend on the physical properties of the arterial tree (stiffness and branching), PWV and the distance to the reflecting sites31. In patients with PH, the PWV in the PA is higher, at about 4.4 m/s5,32, thus the reflected wave reaches the PA in systole, increasing the systolic pressure. The relative increase in pressure amplitude (P) is a measure of the magnitude of the reflected pressure wave (Fig. 3).
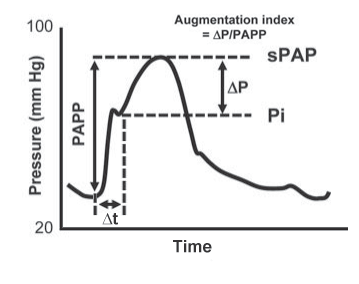
Figure 3. The pressure waveform of the PA in PH. PAPP=pulmonary artery pulse pressure; sPAP = systolic pulmonary artery pressure; Pi=pulmonary artery pressure at the inflection point (at the beginning of the refl ected wave); ΔP=sPAP-Pi (the relative increase in pressure due to wave reflection); Δt= time between the beginning of systole and the beginning of the reflected wave. (reproduced with permission from 26: Champion HC, Michelakis ED, Hassoun PM. Comprehensive invasive and noninvasive approach to the right ventricle-pulmonary circulation unit: state of the art and clinical and research implications. Circulation 2009;120(11):992-1007).
The ratio P/PP defines the augmentation index (AI), which normalizes the pressure rise due to wave reflection for PP. Another important parameter is the time between the beginning of systole and the beginning of the reflected wave (t). One study showed that in patients with chronic thromboembolic pulmonary hypertension (CTEPH) wave reflection is earlier and enhanced compared with patients with idiopathic PAH (iPAH), since CTEPH involves predominantly proximal arteries, while in iPAH, the pathological lesions are mainly located in distal arteries. This finding was expressed by a shorter t and a higher AI in patients with CTEPH33. Moreover, the extent and timing of wave reflection could have an unfavourable effect on ventricular-arterial coupling, loading the stillejecting ventricle.
B. 4. Pulmonary artery stiffness parameters
Several parameters for evaluating pulmonary artery stiffness have been defined and measured (Table 1 and 2). Capacitance is a measure of the ability of arterial vessels to expand during systole and to recoil during diastole. A high capacitance arterial tree lowers the systolic pressure by storing blood during systole and increases diastolic pressure by recoiling during diastole, with 2 effects: damping the pressure variations of the ventricle and supplying constant peripheral blood flow during the entire cardiac cycle9. Most methods of estimating arterial compliance are based on the arterial windkessel method. Parameter optimizations of two- and three-element windkessel models have been developed for this purpose. The three-element windkessel model, which comprises total peripheral resistance, characteristic imped ance and total arterial compliance, leads to signifi cant overestimation of arterial compliance. Moreover, this method is unreliable when wave reflections are small34. The two-element windkessel model, consisting only of total peripheral resistance and total arterial compliance, has lead to three methods of calculating arterial compliance. Two of them, the diastolic decay method and the area method, require measurement of pressure curves and can be applied only when there is no flow in diastole, thus being unsuited for patients with pulmonary hypertension, that present in a vast majority of cases with pulmonary regurgitation35. The third method based on the two-element windkessel model is the pulse pressure (PP) method, which is a reliable method for calculating total arterial compliance in the systemic36 and pulmonary arterial tree34 and does not require detailed pressure curves or no reverse diastolic flow, being applicable for patients with pulmonary hypertension. On the other hand, this method requires the measurement of fl ow curves, vascular resistance and PP35. Since a compliant arterial system dampens ventricular pressure variations, it has been shown that the higher the compliance, the lower the PP is both in the systemic37 and in the pulmonary circulation14. Because PP is mainly determined by stroke volume (SV) and arterial compliance, another method of estimating arterial compliance is the ratio of SV to PP, reflecting total rather than local arterial compliance. This method is also known to overestimate total compliance, since it does not consider outfl ow from the arterial tree during the ejection phase. Nevertheless, this equation has been shown to have a good correlation with the PP method34 and it has been validated in the systemic38 and pulmonary circulation34. Although in one study compliance has been shown to be altered in patients with exercise-induced PH with out overt PAP changes at rest13, it remains unclear if these findings predict the development of PH at rest, especially since the diagnosis of exercise-induced PH is still a matter of controversy39. Moreover, the causal relationship between stiffness and pressure is still unknown; it is unclear if stiffness increases only secondary to increased arterial pressure or due to pressure independent changes of the arterial wall or both. Still, the finding of altered compliance in the absence of PH at rest and moreover, the fact that higher distending pressures do not fully account for the increased stiffness as evaluated by the stiffness index , may suggest a role of altered intrinsic elastic properties of the PA in the early stages of PH and thus worsening PA stiffness among the first physiopathological manifestations of PH13.
Table 1. Parameters of arterial stiffness
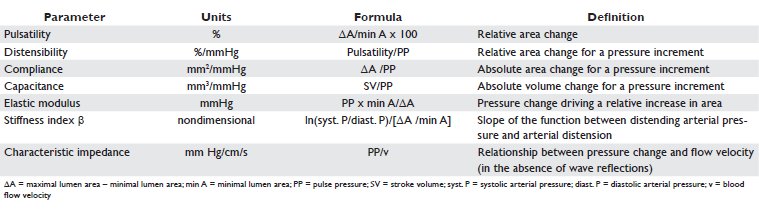
Table 2. Reference values of parameters of arterial stiffness for the pulmonary artery
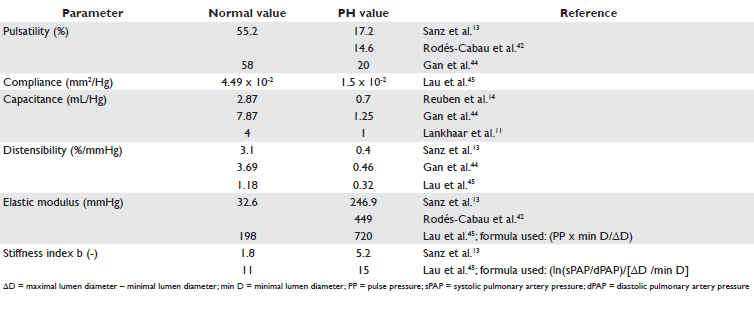
From a clinical perspective, PA stiffness may contribute to the disconnect between the severity of hemodynamic abnormalities and treatment response to vasodilator drugs40. Furthermore, PA distensibility has been shown to predict functional capacity in patients with PAH, as assessed by the 6-minute walk test41. More over, several indices of pulmonary stiffness have been shown to have prognostic value in PH. In one study, decreased pulsatility and increased elastic modulus of distal pulmonary arteries in patients with PAH were associated with mortality at follow-up42. In another study of patients with PAH, pulmonary capacitance, as assessed by the ratio of SV/PP, was a better predictor of mortality than PVR, right atrial pressure, cardiac index, 6-minute walk distance and National Institutes of Health 4-year predicted survival equation43. Another study showed that PA pulsatility was a better marker of death than PVR, right atrial pressure, or 6-minute walk distance44.
C. General aspects of right ventricular-arterial coupling in pulmonary hypertension
RV function is a major determinant of clinical presentation and prognosis in PH, with RV failure being the main cause of death46. The RV is thin-walled compared to the LV, with approximately one-fourth of LV mass47, but due to the low impedance and high compliance of pulmonary circulation it pumps the same volume of blood at the same rate as the LV. Therefore, the RV is especially challenged when it has to adapt to up to 4-5 fold increased chronic afterload, while RV volume overload is better tolerated48. In RV pressure overload, initially adaptive mechanisms come into play, with myocardial hypertrophy preserving RV systolic function and normalising wall stress and with preserved RVA coupling. Over time however RV dilation and systolic dysfunction occur, with RVA uncoupling, RV failure and eventually death48. RVA coupling is best quantifi ed by the relationship between end-systolic or maximal ventricular elastance (Ees or Emax) and effective arterial elastance (Ea), both expressed in the time domain using pressure-volume curves. Ees, the slope of the end-systolic pressure (ESP) – volume relationship, initially characterized for the LV49,50 and afterwards for the RV51, is a measure of ventricular contractile state, being nearly independent of changes in preload, afterload, and heart rate (Fig. 4). Moreover, Ees integrates besides intrinsic ventricular contractility the modulating effects of geometric and biochemical properties of the ventricle52. Ees can be calculated as ESP/[end-systolic volume (ESV)-V0], where V0 is the volume corresponding to the x-axis intercept of the ESP-V linear relationship and represents the theoretical volume of the unloaded ventricle49,53. A common assumption is that V0 is negligible compared with ESV, thus the equation becomes Ees = ESP/ESV54. On the other hand, Ea is an integrative index of arte rial load, encompassing its principal elements: peripheral resistance, total arterial compliance, characteristic impedance and systolic and diastolic time intervals55. Despite its broad spectrum, Ea is dominated by nonpulsatile load56. This parameter is determined from pressure-volume loops as the negative slope of the line between the end-diastolic volume (EDV) and ESP points and can be approximated by the ratio of ESP to SV for the LV57, while for the RV the equation becomes Ea = [ESP-pulmonary capillary wedge pressure (PCWP)]/SV58. The coupling between the ventricle and the respective artery is expressed by the relation of the two elastances and calculated as Ees/Ea or Ea/Ees54. Effective coupling can be defi ned using several criteria, the most important being optimal external work and optimal energetic efficiency of the heart. Numerically, it has been predicted57 and experimentally observed that near optimal work and effi ciency are achieved when the Ea/Ees ratio is between 0.6 and 1.2 for the LV52,59, range normally maintained in various physiological conditions. For the RV normal values at rest between 0.37 and 0.7 have been reported60-63, with optimal mechanical coupling occurring when the ratio is 1, while the optimal energy transfer from the RV to the pulmonary arterial tree occurs at ratios of 0,67-12. Technically, the use of pressure-volume curves with varying load is more challenging for the RV than for the LV. Furthermore, its crescent shape makes accurate instantaneous RV volume measurements more diffi cult26. Moreover, the almost ”triangular” shape of the pressure-volume curve due to the ongoing ventricular ejection despite a signifi cant drop in pressure makes the defi nition of the maximal ventricular elastance more challenging, since it occurs before the end of systole in the RV64. More recently a single-beat method has been developed, which bypasses the challenging load alterations and the difficulty of measuring instantaneous RV volume in vivo65 (Fig. 4). Furthermore, hybrid methods have been developed, which use invasively measured pressures and non-invasively determined volumes, either with MRI61 or with 3D real-time echocardiography66. Recently, a completely non-invasive method of assessing RV-arterial (RVA) coupling using MRI has been described, with excellent correlation and good agreement with invasively determined coupling, albeit with a systematical overestimation, but with equivalent patterns of progression across disease stages in PH. In this study a simplified formula has been used, by assuming V0 negligible in the equation for Ees and PCWP negligible in the equation for Ea and as such Ea/Ees = ESP/SV/ESP/ESV = ESV/SV62. In an animal model of PH induced by acute pulmonary embolism with gradual PAP increases, Ea increased progressively, while Ees increased initially, but failed to increase any further. As a result, Ea/Ees initially increased to values associated with optimal stroke work, suggesting that RV contractile reserve had been recruited to a point of optimal coupling, but submaximal efficiency, whereas in later stages the ratio increased further, suggesting RVA uncoupling and RV failure67. These findings were confirmed in a larger human study of patients with PH of several etiologies, where Ea increased linearly with advancing severity of PH, while Ees increased initially, but failed to increase further and tended even to decrease in more advanced stages. Thus, RVA coupling ratio was relatively maintained in earlier stages of PH, but increased markedly from a bout 0.35 till 2.85 across quartiles of PVR index62. In another study, patients with CTEPH and patients with chronic thromboembolic disease without PH (CTED) were analyzed using conductance catheterderived pressure-volume loops with the single-beat method. Similarly to the previous study, patients with CTEPH had higher Ees compared to CTED and controls, but with Ea/Ees suggesting RVA uncoupling, with increased values at 1.67 compared to 0.69 in controls and 0.79 in CTED63. In a small subset of a study that included patients with systemic sclerosis-associated PAH (SSPAH) and patients with iPAH, RV pressurevolume loops were measured and Ees, Ea and Ea/Ees were derived. Patients with SSPAH and with iPAH had similar steady and pulsatile afterload, with similar PVR and PA compliance, but with decreased Ees and increased Ea/Ees ratio for patients with SSPAH compared with iPAH, suggesting that the impaired prognosis in this group compared to iPAH is associated with a worse RV function and impaired RVA coupling at similar levels of afterload68. The impact of afterload on RV function differs between steady and pulsatile afterload, as has been shown in another study that assessed PA stiffness and its association with RV parameters. In this study, patients with PH were analyzed by RHC and MRI; PA elasticity, distensibility, capacitance and stiffness index were independently associated with RV ejection fraction, mass index, end-systolic volume index and stroke work index, after adjusting for age, sex and PVR index. Moreover, the relative contributions of these parameters of PA stiffness for RV stroke work index were 1.2 x to 18 higher than those of PVR index, suggesting that RV adaptation to chronic pressure afterload is more strongly related to pulsatile than to steady afterload69. Another study found similar results in a group of patients with atrial septal defect, demonstrating PA stiffness to be the best correlate of RV function as assessed by the myocardial performance index, better than invasively determined PAPs or PVR70. Another study showed that, despite a reduction in PVR after PAH-targeted therapy, in 25% of patients RV function deteriorates further and is associated with a poor outcome. The cause is not fully known, but it is suggested that unfavourable RVA coupling might play a role71. Interestingly, a recent study showed in an animal model of chronic RV pressure overload that echocardiographic indices of RV function, such as the RV myocardial performance index and the myocardial acceleration during isovolumic contraction are better correlated with RVA coupling ratio than with Ees72. RVA coupling measured with the Ea/Ees ratio has been assessed in several animal models of PH with and without RV failure and in models with persistent RV failure after transient pulmonary artery banding and pharmacological interventions. In summary, Ea increased in all models, while RVA coupling tended to be maintained by an adaptive increase in Ees in models such as hypoxic exposure73,74, in early sepsis75, in acute embolism74, in PA banding74 or a few months of aortopulmonary shunting76 associated with only a moderate increase in mean PAP. Prolonged mechanical stress, such as with 6 months of overcirculation in piglets77, in late sepsis75 or in monocrotaline-induced PH78 may cause uncoupling of the RV from the pulmonary circulation, with insufficiently increased Ees or even decreased Ees and consequently increased Ea/Ees. A general trend in the mentioned studies is that increased pulmonary arterial obstruction (for instance pulmonary stenosis (PS) or pulmonary artery banding) allows for longer preservation of RVA coupling when compared to situations where the PVR is increased because of pulmonary vascular diseases2,79. Superior RV function in this setting has been confirmed in a human study that included patients with PS and with PAH at the same level of RV pressure overload and found better RV function parameters and less RV dilation in patients with PS80.
D. Clinical implications of right ventricular-arterial coupling assessment
The effect of several pharmacological interventions on RVA coupling have been studied in these animal models, with beneficial or deleterious effects. Milrinone81 and chronic administration of bisoprolol82 have shown favourable effects on RVA coupling, because of positive inotropic effects with increased Ees and no effect on Ea, with consequent decrease in Ea/Ees ratio. Combined positive inotropy (increased Ees) and vasodila tion (decreased Ea), with subsequent decrease in Ea/Ees have shown low-dose dobutamine, low-dose norepinephrine (less pronounced effects than dobutamine) 83, levosimendan84 and sildenafi l, the latter in monocrotaline-induced PH85. In the setting of hypoxia, imsildenafil has shown favourable effects on RVA coupling through vasodilating and no inotropic effects86, similar to acute epoprostenol administration or inhaled nitric oxide87. Acute administration of propranolol had deleteri ous effects on RVA coupling, with negative inotropic effects and pulmonary vasoconstriction during acute hypoxia65. The importance of assessing RVA coupling has been recently underlined by a human study which included patients with PH, in which RVA coupling remained the only independent predictor of transplantation-free survival after controlling for right atrial pressure, mean PAP and SV88. Another important aspect of RVA coupling is the assessment of RV contractile reserve, which may unmask borderline or latent resting uncoupling. This topic is an emerging research area; stress testing using exercise or dobutamine have been used. In one study exercise-induced increase in systolic RV pressure, as estimated from the tricuspid regurgitant jet, has been shown to be a strong predictor of survival in patients with PH89. Dobutamine stress has the advantage of tech nically easier imaging and has been demonstrated in one study to reveal subclinical reduction in RV contractile reserve in patients with PAH and preserved RV function at rest, as assessed by the change in TAPSE and tricuspid annular systolic velocity compared to controls90. In an animal model resting RVA coupling has been shown to correlate with RV reserve as assessed by dobutamine stress echocardiography91. Assessment of RVA coupling adds to the physiopathological understanding and the clinical and therapeutic assessment in PH. Further studies are needed to better understand the mechanisms that lead from efficient RVA coupling to uncoupling and thus to translate these findings into efficient treatment for PH patients.
Funding: This work was supported by the Sectoral Operational Programme Human Resources Development (SOP HRD), financed from the European Social Fund and by the Romanian Government under the contract number POSDRU/159/1.5/S/132395 (EP) and by the UMF “Carol Davila” “Young researchers projects”, contract number 23348/04.11.2013 (RJ).
Conflict of interest: none stated.
References
1. Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, et al. Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation 2006;114(17):1883-91.
2. Naeije R, Brimioulle S, Dewachter L. Biomechanics of the right ventricle in health and disease (2013 Grover Conference series). Pulm Circ 2014;4(3):395-406.
3. Kussmaul WG, Noordergraaf A, Laskey WK. Right ventricular-pulmonary arterial interactions. Ann Biomed Eng 1992;20(1):63-80.
4. O’Rourke MF. Vascular impedance in studies of arterial and cardiac function. Physiol Rev 1982;62(2):570-623.
5. Milnor WR, Conti CR, Lewis KB, O’Rourke MF. Pulmonary arterial pulse wave velocity and impedance in man. Circ Res 1969;25(6):637-49.
6. Saouti N, Westerhof N, Helderman F, Marcus JT, Boonstra A, Postmus PE, et al. Right ventricular oscillatory power is a constant fraction of total power irrespective of pulmonary artery pressure. Am J Resp Crit Care Med 2010;182(10):1315-20.
7. O’Rourke MF. Steady and pulsatile energy losses in the systemic circulation under normal conditions and in simulated arterial disease. Cardiovasc Res 1967;1(4):313-26.
8. Nichols WW, O’Rourke MF, Avolio AP, Yaginuma T, Pepine CJ, Conti CR. Ventricular/vascular interaction in patients with mild systemic hypertension and normal peripheral resistance. Circulation 1986;74(3):455-62.
9. Saouti N, Westerhof N, Postmus PE, Vonk-Noordegraaf A. The arterial load in pulmonary hypertension. Eur Respir Rev 2010;19(117):197-203.
10. Dawson CA, Rickaby DA, Linehan JH, Bronikowski TA. Distributions of vascular volume and compliance in the lung. J Appl Physiol 1988;64(1):266-73.
11. Lankhaar JW, Westerhof N, Faes TJ, Marques KM, Marcus JT, Postmus PE, et al. Quantifi cation of right ventricular afterload in patients with and without pulmonary hypertension. Am J Physiol Heart Circ Physiol 2006;291(4):H1731-7.
12. Lankhaar JW, Westerhof N, Faes TJ, Gan CT, Marques KM, Boonstra A, et al. Pulmonary vascular resistance and compliance stay inversely related during treatment of pulmonary hypertension. Eur Heart J 2008;29(13):1688-95.
13. Sanz J, Kariisa M, Dellegrottaglie S, Prat-Gonzalez S, Garcia MJ, Fuster V, et al. Evaluation of pulmonary artery stiffness in pulmonary hypertension with cardiac magnetic resonance. JACC Cardiovasc Imaging 2009;2(3):286-95.
14. Reuben SR. Compliance of the human pulmonary arterial system in disease. Circ Res 1971;29(1):40-50.
15. Haimovici JB, Trotman-Dickenson B, Halpern EF, Dec GW, Ginns LC, Shepard JA, et al. Relationship between pulmonary artery diameter at computed tomography and pulmonary artery pressures at rightsided heart catheterization. Massachusetts General Hospital Lung Transplantation Program. Acad Radiol 1997;4(5):327-34.
16. Kuriyama K, Gamsu G, Stern RG, Cann CE, Herfkens RJ, Brundage BH. CT-determined pulmonary artery diameters in predicting pulmonary hypertension. Invest Radiol 1984;19(1):16-22.
17. Frank H, Globits S, Glogar D, Neuhold A, Kneussl M, Mlczoch J. Detection and quantification of pulmonary artery hypertension with MR imaging: results in 23 patients. AJR Am J Roentgenol 1993;161(1):27-31.
18. Murray TI, Boxt LM, Katz J, Reagan K, Barst RJ. Estimation of pulmonary artery pressure in patients with primary pulmonary hypertension by quantitative analysis of magnetic resonance images. J Thorac Imaging 1994;9(3):198-204.
19. Ng CS, Wells AU, Padley SP. A CT sign of chronic pulmonary arterial hypertension: the ratio of main pulmonary artery to aortic diameter. J Thorac Imaging 1999;14(4):270-8.
20. Boerrigter B, Mauritz GJ, Marcus JT, Helderman F, Postmus PE, Westerhof N, et al. Progressive dilatation of the main pulmonary artery is a characteristic of pulmonary arterial hypertension and is not related to changes in pressure. Chest 2010;138(6):1395-401.
21. Kobs RW, Chesler NC. The mechanobiology of pulmonary vascular remodeling in the congenital absence of eNOS. Biomech Model Mechanobiol 2006;5(4):217-25.
22. Lammers SR, Kao PH, Qi HJ, Hunter K, Lanning C, Albietz J, et al. Changes in the structure-function relationship of elastin and its imvopact on the proximal pulmonary arterial mechanics of hypertensive calves. Am J Physiol Heart Circ Physiol 2008;295(4):H1451-9.
23. Huez S, Brimioulle S, Naeije R, Vachiery JL. Feasibility of routine pulmonary arterial impedance measurements in pulmonary hypertension. Chest 2004;125(6):2121-8.
24. Hunter KS, Lee PF, Lanning CJ, Ivy DD, Kirby KS, Claussen LR, et al. Pulmonary vascular input impedance is a combined measure of pulmonary vascular resistance and stiffness and predicts clinical outcomes better than pulmonary vascular resistance alone in pediatric patients with pulmonary hypertension. Am Heart J 2008;155(1):166-74.
25. Bergel DH, Milnor WR. Pulmonary Vascular Impedance in the Dog. Circ Res 1965;16:401-15.
26. Champion HC, Michelakis ED, Hassoun PM. Comprehensive invasive and noninvasive approach to the right ventricle-pulmonary circulation unit: state of the art and clinical and research implications. Circulation 2009;120(11):992-1007.
27. Murgo JP, Westerhof N, Giolma JP, Altobelli SA. Aortic input impedance in normal man: relationship to pressure wave forms. Circulation 1980;62(1):105-16.
28. Merillon JP, Motte G, Masquet C, Guiomard A, Baudouy Y, Gourgon R. [Evaluation of static elasticity and characteristic impedence of the aorta. Their relationships with age, aortic pressure and ventricular ejection resistance]. Arch Mal Coeur Vaiss 1976;69(7):653-9.
29. Latham RD, Westerhof N, Sipkema P, Rubal BJ, Reuderink P, Murgo JP. Regional wave travel and refl ections along the human aorta: a study with six simultaneous micromanometric pressures. Circulation 1985;72(6):1257-69.
30. van den Bos GC, Westerhof N, Randall OS. Pulse wave reflection: can it explain the differences between systemic and pulmonary pressure and flow waves? A study in dogs. Circ Res 1982;51(4):479-85.
31. Townsend RR, Wilkinson IB, Schiffrin EL, Avolio AP, Chirinos JA, Cockcroft JR, et al. Recommendations for Improving and Standardizing Vascular Research on Arterial Stiffness: A Scientific Statement From the American Heart Association. Hypertension 2015.
32. Caro CG, Harrison GK. Observations on pulse wave velocity and pulsatile blood pressure in the human pulmonary circulation. Clin Sci 1962;23:317-29.
33. Castelain V, Herve P, Lecarpentier Y, Duroux P, Simonneau G, Chemla D. Pulmonary artery pulse pressure and wave refl ection in chronic pulmonary thromboembolism and primary pulmonary hypertension. J Am Coll Cardiol 2001;37(4):1085-92.
34. Segers P, Brimioulle S, Stergiopulos N, Westerhof N, Naeije R, Maggiorini M, et al. Pulmonary arterial compliance in dogs and pigs: the three-element windkessel model revisited. Am J Physiol 1999;277(2 Pt 2):H725-31.
35. Muthurangu V, Atkinson D, Sermesant M, Miquel ME, Hegde S, Johnson R, et al. Measurement of total pulmonary arterial compliance using invasive pressure monitoring and MR flow quantification during MR-guided cardiac catheterization. Am J Physiol Heart Circ Physiol 2005;289(3):H1301-6.
36. Stergiopulos N, Meister JJ, Westerhof N. Simple and accurate way for estimating total and segmental arterial compliance: the pulse pressure method. Ann Biomed Eng 1994;22(4):392-7.
37. Ferguson JJ, 3rd, Randall OS. Hemodynamic correlates of arterial compliance. Cathet Cardiovasc Diagn 1986;12(6):376-80.
38. Henriksen JH, Fuglsang S, Bendtsen F, Christensen E, Moller S. Arterial compliance in patients with cirrhosis: stroke volume-pulse pressure ratio as simplified index. Am J Physiol Gastrointest Liver Physiol 2001;280(4):G584-94.
39. Kovacs G, Berghold A, Scheidl S, Olschewski H. Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J 2009;34(4):888-94.
40. Ben-Yehuda O, Barnett C. Magnetic resonance assessment of pulmonary artery compliance: a promising diagnostic and prognostic tool in pulmonary hypertension? JACC Cardiovasc Imaging 2009;2(3):296-8.
41. Kang KW, Chang HJ, Kim YJ, Choi BW, Lee HS, Yang WI, et al. Cardiac magnetic resonance imaging-derived pulmonary artery distensibility index correlates with pulmonary artery stiffness and predicts functional capacity in patients with pulmonary arterial hypertension. Circ J 2011;75(9):2244-51.
42. Rodes-Cabau J, Domingo E, Roman A, Majo J, Lara B, Padilla F, et al. Intravascular ultrasound of the elastic pulmonary arteries: a new approach for the evaluation of primary pulmonary hypertension. Heart 2003;89(3):311-5.
43. Mahapatra S, Nishimura RA, Oh JK, McGoon MD. The prognostic value of pulmonary vascular capacitance determined by Doppler echocardiography in patients with pulmonary arterial hypertension. J Am Soc Echocardiogr 2006;19(8):1045-50.
44. Gan CT, Lankhaar JW, Westerhof N, Marcus JT, Becker A, Twisk JW, et al. Noninvasively assessed pulmonary artery stiffness predicts mortality in pulmonary arterial hypertension. Chest 2007;132(6):1906-12.
45. Lau EM, Iyer N, Ilsar R, Bailey BP, Adams MR, Celermajer DS. Abnormal pulmonary artery stiffness in pulmonary arterial hypertension: in vivo study with intravascular ultrasound. PloS One 2012;7(3):e33331.
46. Galie N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009;30(20):2493-537.
47. Sandstede J, Lipke C, Beer M, Hofmann S, Pabst T, Kenn W, et al. Ageand gender-specifi c differences in left and right ventricular cardiac function and mass determined by cine magnetic resonance imaging. Eur Radiol 2000;10(3):438-42.
48. Jurcut R, Giusca S, La Gerche A, Vasile S, Ginghina C, Voigt JU. The echocardiographic assessment of the right ventricle: what to do in 2010? Eur J Echocardiogr 2010;11(2):81-96.
49. Suga H, Sagawa K, Shoukas AA. Load independence of the instantaneous pressure-volume ratio of the canine left ventricle and effects of epinephrine and heart rate on the ratio. Circ Res 1973;32(3):314-22.
50. Suga H, Sagawa K. Instantaneous pressure-volume relationships and their ratio in the excised, supported canine left ventricle. Circ Res 1974;35(1):117-26.
51. Maughan WL, Shoukas AA, Sagawa K, Weisfeldt ML. Instantaneous pressure-volume relationship of the canine right ventricle. Circ Res 1979;44(3):309-15.
52. Borlaug BA, Kass DA. Ventricular-vascular interaction in heart failure. Heart Fail Clin 2008;4(1):23-36.
53. Chantler PD, Lakatta EG, Najjar SS. Arterial-ventricular coupling: mechanistic insights into cardiovascular performance at rest and during exercise. J Appl Physiol 008;105(4):1342-51.
54. Sunagawa K, Maughan WL, Sagawa K. Optimal arterial resistance for the maximal stroke work studied in isolated canine left ventricle. Circ Res1985;56(4):586-95.
55. Kelly RP, Ting CT, Yang TM, Liu CP, Maughan WL, Chang MS, et al. Effective arterial elastance as index of arterial vascular load in humans. Circ 1992;86(2):513-21.
56. Chirinos JA, Rietzschel ER, Shiva-Kumar P, De Buyzere ML, Zamani P, Claessens T, et al. Effective arterial elastance is insensitive to pulsatile arterial load. Hypertension 2014;64(5):1022-31.
57. Sunagawa K, Maughan WL, Burkhoff D, Sagawa K. Left ventricular interaction with arterial load studied in isolated canine ventricle. Am J Physiol. 1983;245(5 Pt 1):H773-80.
58. Morimont P, Lambermont B, Ghuysen A, Gerard P, Kolh P, Lancellotti P, et al. Effective arterial elastance as an index of pulmonary vascular load. Am J Physiol Heart Circ Physiol 2008;294(6):H2736-42.
59. Chen CH, Nakayama M, Nevo E, Fetics BJ, Maughan WL, Kass DA. Coupled systolic-ventricular and vascular stiffening with age: implications for pressure regulation and cardiac reserve in the elderly. J Am Coll Cardiol 1998;32(5):1221-7.
60. Spruijt OA, de Man FS, Groepenhoff H, Oosterveer F, Westerhof N, Vonk-Noordegraaf A, et al. The effects of exercise on right ventricular contractility and right ventricular-arterial coupling in pulmonary hypertension. Am J Respir Crit Care Med 2015;191(9):1050-7.
61. Kuehne T, Yilmaz S, Steendijk P, Moore P, Groenink M, Saaed M, et al. Magnetic resonance imaging analysis of right ventricular pressure-volume loops: in vivo validation and clinical application in patients with pulmonary hypertension. Circulation 2004;110(14):2010-6.
62. Sanz J, Garcia-Alvarez A, Fernandez-Friera L, Nair A, Mirelis JG, Sawit ST, et al. Right ventriculo-arterial coupling in pulmonary hypertension: a magnetic resonance study. Heart 2012;98(3):238-43.
63. McCabe C, White PA, Hoole SP, Axell RG, Priest AN, Gopalan D, et al. Right ventricular dysfunction in chronic thromboembolic obstruction of the pulmonary artery: a pressure-volume study using the conductance catheter. J Appl Physiol 2014;116(4):355-63.
64. Giusca S, Jurcut R, Ginghina C, Voigt JU. The right ventricle: anatomy, physiology and functional assessment. Acta cardiol 2010;65(1):67-77.
65. Brimioulle S, Wauthy P, Ewalenko P, Rondelet B, Vermeulen F, Kerbaul F, et al. Single-beat estimation of right ventricular end-systolic pressure-volume relationship. Am J Physiol Heart Circ Physiol 2003;284(5):H1625-30.
66. Herberg U, Gatzweiler E, Breuer T, Breuer J. Ventricular pressurevolume loops obtained by 3D real-time echocardiography and mini pressure wire-a feasibility study. Clin Res Cardiol 2013;102(6):427-38.
67. Ghuysen A, Lambermont B, Kolh P, Tchana-Sato V, Magis D, Gerard P, et al. Alteration of right ventricular-pulmonary vascular coupling in a porcine model of progressive pressure overloading. Shock 2008; 29(2):197-204.
68. Tedford RJ, Mudd JO, Girgis RE, Mathai SC, Zaiman AL, Housten-Harris T, et al. Right ventricular dysfunction in systemic sclerosis-associated pulmonary arterial hypertension. Circ Heart Fail 2013;6(5):953-63.
69. Stevens GR, Garcia-Alvarez A, Sahni S, Garcia MJ, Fuster V, Sanz J. RV dysfunction in pulmonary hypertension is independently related to pulmonary artery stiffness. JACC Cardiovasc Imaging 2012;5(4):378-87.
70. Gorgulu S, Eren M, Uslu N, Ozer O, Nurkalem Z. The determinants of right ventricular function in patients with atrial septal defect. Int J Cardiol 2006;111(1):127-30.
71. van de Veerdonk MC, Kind T, Marcus JT, Mauritz GJ, Heymans MW, Bogaard HJ, et al. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol 2011;58(24):2511-9.
72. Guihaire J, Haddad F, Boulate D, Decante B, Denault AY, Wu J, et al. Non-invasive indices of right ventricular function are markers of ventricular-arterial coupling rather than ventricular contractility: insights from a porcine model of chronic pressure overload. Eur Heart J Cardiovasc Imaging 2013;14(12):1140-9.
73. Kerbaul F, Rondelet B, Motte S, Fesler P, Hubloue I, Ewalenko P, et al. Isofl urane and desfl urane impair right ventricular-pulmonary arterial coupling in dogs. Anesthesiology 2004;101(6):1357-62.
74. Wauthy P, Pagnamenta A, Vassalli F, Naeije R, Brimioulle S. Right ventricular adaptation to pulmonary hypertension: an interspecies comparison. Am J Physiol Heart Circ Physiol 2004;286(4):H1441-7.
75. Lambermont B, Ghuysen A, Kolh P, Tchana-Sato V, Segers P, Gerard P, et al. Effects of endotoxic shock on right ventricular systolic function and mechanical efficiency. Cardiovasc Res 2003;59(2):412-8.
76. Rondelet B, Kerbaul F, Motte S, van Beneden R, Remmelink M, Brimioulle S, et al. Bosentan for the prevention of overcirculation-in duced experimental pulmonary arterial hypertension. Circulation 2003;107(9):1329-35.
77. Rondelet B, Dewachter C, Kerbaul F, Kang X, Fesler P, Brimioulle S, et l. Prolonged overcirculation-induced pulmonary arterial hypertension as a cause of right ventricular failure. Eur Heart J 2012;33(8):1017-26.
78. Gomez-Arroyo JG, Farkas L, Alhussaini AA, Farkas D, Kraskauskas D, Voelkel NF, et al. The monocrotaline model of pulmonary hypertension in perspective. Am J Physiol Lung Cell Mol Physiol 2012;302(4):L363-9.
79. Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, et al. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 2009;120(20):1951-60.
80. Jurcut R, Giusca S, Ticulescu R, Popa E, Amzulescu MS, Ghiorghiu I, et al. Different patterns of adaptation of the right ventricle to pressure overload: a comparison between pulmonary hypertension and pulmonary stenosis. J Am Soc Echocardiogr 2011;24(10):1109-17.
81. Pagnamenta A, Dewachter C, McEntee K, Fesler P, Brimioulle S, Naeije R. Early right ventriculo-arterial uncoupling in borderline pulmonary hypertension on experimental heart failure. J Appl Physiol 2010;109(4):1080-5.
82. de Man FS, Handoko ML, van Ballegoij JJ, Schalij I, Bogaards SJ, Postmus PE, et al. Bisoprolol delays progression towards right heart failure in experimental pulmonary hypertension. Circ Heart Fail 2012;5(1):97-105.
83. Kerbaul F, Rondelet B, Motte S, Fesler P, Hubloue I, Ewalenko P, et al. Effects of norepinephrine and dobutamine on pressure load-induced right ventricular failure. Crit Care Med 2004;32(4):1035-40.
84. Missant C, Rex S, Segers P, Wouters PF. Levosimendan improves right ventriculovascular coupling in a porcine model of right ventricular dysfunction. Crit Care Med 2007;35(3):707-15.
85. Borgdorff MA, Bartelds B, Dickinson MG, Boersma B, Weij M, Zandvoort A, et al. Sildenafi l enhances systolic adaptation, but does not prevent diastolic dysfunction, in the pressure-loaded right ventricle. Eur J Heart Fail 2012;14(9):1067-74.
86. Fesler P, Pagnamenta A, Rondelet B, Kerbaul F, Naeije R. Effects of sildenafil on hypoxic pulmonary vascular function in dogs. J Appl Physiol 2006;101(4):1085-90.
87. Wauthy P, Abdel Kafi S, Mooi WJ, Naeije R, Brimioulle S. Inhaled nitric oxide versus prostacyclin in chronic shunt-induced pulmonary hypertension. J Thorac Cardiovasc Surg 2003;126(5):1434-41.
88. Vanderpool RR, Pinsky MR, Naeije R, Deible C, Kosaraju V, Bunner C, et al. RV-pulmonary arterial coupling predicts outcome in patients referred for pulmonary hypertension. Heart 2015;101(1):37-43.
89. Grunig E, Tiede H, Enyimayew EO, Ehlken N, Seyfarth HJ, Bossone E, et al. Assessment and prognostic relevance of right ventricular contractile reserve in patients with severe pulmonary hypertension. Circulation 2013;128(18):2005-15.
90. Sharma T, Lau EM, Choudhary P, Torzillo PJ, Munoz PA, Simmons LR, et al. Dobutamine stress for evaluation of right ventricular reserve in pulmonary arterial hypertension. Eur Respir J 2015;45(3):700-8.
91. Guihaire J, Haddad F, Noly PE, Boulate D, Decante B, Dartevelle P, et al. Right ventricular reserve in a piglet model of chronic pulmonary hypertension. Eur Respir J 2015;45(3):709-17.
 This work is licensed under a
This work is licensed under a