Ioan Mircea Coman1, Simona Bațaga2, Miron Bogdan1, Mircea Diculescu1, Cristina Gavrilă3, Ioan Manițiu4, Zeno Sparchez5, Ioan Țilea2
1 University of Medicine and Pharmacy “Carol Davila”, Bucharest
2 University of Medicine and Pharmacy Tîrgu Mureș, Tîrgu Mureș
3 Emergency Institute for Cardiovascular Diseases „Prof. Dr. C.C Iliescu”, Bucharest
4 University of Medicine and Pharmacy “Lucian Blaga”, Sibiu
5 University of Medicine and Pharmacy “Iuliu Hațieganu”, Cluj Napoca
GENERALS
Portopulmonary hypertension (PPHT) is a known complication, infrequent, but serious, of patients with portal hypertension (HT), primarily of those with cirrhosis, occurring in 5-10% of patients assessed for liver transplantation (1). Some risk factors have been associated with the development of PPHT: of these, female gender and autoimmune hepatitis have a greater risk, while patients with viral hepatitis C have a lower risk of developing PPHT (2). PPHT may also occur in the Budd Chiari syndrome, extrahepatic portal vein obstruction, portal vein thrombosis, portal cavernoma, etc. The definition of portopulmonary hypertension has been modified over time, in particular with regard to the threshold of pulmonary vascular resistance (PVR). Recently, researchers at the Mayo Clinic (3) proposed new diagnostic criteria of PPHT: (1) Portal hypertension and/or liver disease, based on the clinical diagnosis – ascites, esophageal varices, splenomegaly; (2) Mean pulmonary artery pressure (mPAP)* >25 mmHg; and (3) PVR* >3 Wood units (Wu); and (4) Pulmonary capillary wedge pressure (PCWP)* 12 mmHg, where GTP= mPAP-PCWP. *measurements obtained by cardiac catheterization at rest. Moderately-severe forms of PPHT have a very reserved prognosis (5-year survival rate is 40% in the US REVEAL registry, namely 68% in the French registry) (4.5). Hemodynamic studies confirmed the presence of pulmonary arterial hypertension in 2-6% of patients with portal hypertension (4). This pathology is currently severely underdiagnosed in Romania; from an estimated number of 40,000 – 80,000 patients with cirrhosis, we can estimate that over 1,000 PPHT carriers remain (mostly) unidentified.
The pathophysiological mechanisms remain uncertain. There is no proportionality between the degree of portal hypertension, liver dysfunction and development of PPHT. Recent data of a retrospective study suggest a strong association between the presence of large portosystemic shunts (especially those splenorenal), portal hepatofugal blood flow and PPHT6. It is to be noted that the presence of these shunts was observed to be a criterion for the absence of response to specific vasodilator drug therapy. From a circulatory point of view, the patients with hepatic cirrhosis show a hyperdynamic status manifested by increased heart rate; the differentiation of PPHT from the hepato-pulmonary syndrome is to be noted, characterized by increased flow rates and low pulmonary resistances.
Non-invasive diagnosis of PPHT is difficult due to the nonspecific clinical picture. The main clinical manifestations are dyspnea on exertion or at rest and cyanosis of central type. Confirmation of portal HT is done according to the classic diagnostic criteria (Appendix 1) Routine investigations (cardiopulmonary X-ray, electrocardiogram, biological and functional respiratory samples) are not sufficient for positive diagnosis or exclusion of associated pulmonary hypertension. Echocardiography is currently the most useful noninvasive method. It provides anatomical data (right ventricular dilation and hypertrophy, dilatation of pulmonary artery) and functional data on pulmonary circulation (with reasonable specifi city): systolic/mean pressure in the pulmonary artery, systolic/diastolic function of the right ventricle, pulmonary/tricuspid regurgitation (Annex 2). Its use for screening in all patients with cirrhosis of the liver in the presence of signs of portal hypertension is difficult to achieve in current practice, due to the large number of patients with this pathology and to the risk of overloading the specialized laboratories; but it remains a mandatory diagnostic phase in patients referred for liver transplantation, and in patients with high probability of PPHT – both to confirm the diagnosis and for the follow up (including for predicting treatment efficacy) and prognostic assessment. Sectional imaging (CT, MRI) dedicated for liver and spleen assessment (extended to the bottom of the thorax) can signal right cardiomegaly or the presence of pericardial fluid. BNP and NT-proBNP dosage (as marker of suffering of the right ventricle) has a limited positive predictive value due to their frequent growth in cirrhotic patients without associated PHT. A useful diagnostic algorithm of PPHT should have a good predictive value (negative and positive) to be scientifically justified and economically applicable in practice. The results of non-invasive tests should provide sufficient information to establish a probable diagnosis, to clarify the need for and frequency of subsequent monitoring or the need/time of performing invasive measurements by right cardiac catheterization.
Invasive diagnosis of PPHT is made by cardiac catheterization and it will be reserved to patients with echocardiographic suspicion of PPHT (systolic pressure in the pulmonary artery over 50 mmHg ± dysfunction of right ventricle ± dilation of right cavities). The measured parameters, useful in establishing a diagnosis of certainty consist of mPAP, PCWP, PVR. To be mentioned that for the exclusion of alternative etiologies of PHT and for appropriate calculation of flows and resistances, both right and left cardiac catheterization are necessary. PPHT is considered mild if mPAP mPAP ≥ 45 mmHg (7). It should be noted that the severity of PPHT does not correlate with portal hypertension or with the severity of hepatic disease (8). Vasodilator tests are not indicated in patients with PPHT because calcium blocker medication is not recommended in this group of patients, as it may cause an increase in hepatic venous gradient and worsen portal hypertension. The risk of incidents and mortality of invasive evaluation in centers with expertise in evaluation PHT (9) are very low (complication rate less than 1% and a mortality less than 0.1%).
TREATMENT
PHT specific drug therapy consists of pulmonary vasodilators; it includes drugs that act on the 3 pathways well known to be involved in the development of PAH (pulmonary arterial hypertension). European Guidelines (2009, 2015) and American Guidelines (2009) and the recent World Symposium of Nice (2013) (10) define the indication class and level of evidence for each of the substances, depending on the patient’s functional class. Only part of the drugs listed have published studies in patients with PPHT.
(1) Endothelin pathway: endothelin receptor inhibitors (Bosentan, Ambrisentan, Macitentan). There are several clinical studies on PPHT, proving an improvement in hemodynamic profile; the patients on Ambrisentan medication had unchanged liver tests at 12 months (11), compared with Bosentan, which gave hepatic cytolysis in some patients12;
(2) Nitric oxide pathway – includes phosphodieterase-5 inhibitors (Sildenafi l, Tadalafi l) and Riociguat. Sildenafi l has the advantage of the absence of hepatotoxicity; it has been used in studies both in monotherapy, and in combined therapy (13), with hemodynamic improvement.
(3) Prostaglandins pathway (Epoprostenol, Iloprost, Treprostinil, Selexipag). It includes the most commonly used medication in clinical trials of the pre-transplant stage, very good results with epoprostenol – net hemodynamic improvement, but not major changes in terms of survival (follow up time 15.4 months) (14).
Treatment of RV dysfunction is made according to usual standards.
Anticoagulation is usually contraindicated in this group of pulmonary hypertension due to the high risk of bleeding in patients with underlying liver disease and portal hypertension, especially in patients with a history of gastrointestinal bleeding. PPHT is a serious problem when we refer to liver transplantation. Mild PPHT, as defined above, is no additional risk, in contrast to the moderate PPHT, which is associated with moderate PPHT, which is associated with perioperative mortality of 50-80% (4), and at values of mPAP above 50 mmHg, mortality approaches 100% (15). When values of mPAP are recorded between 35-50 mmHg, intensive vasodilator treatment is recommended for 3-6 months, and if treatment to therapy is positive, liver transplantation is performed. The evolution of pulmonary hypertension after liver transplantation is generally favorable, but some patients may have a stationary evolution, thus requiring the continuation of specifi c pulmonary vasodilator treat ment. Diagnosis and treatment of portopulmonary hypertension remains a complex one, which is why patients that are suspected as having PPHT should be referred to centers of expertise. The initiative group proposes the following algorithm for diagnosis and treatment:
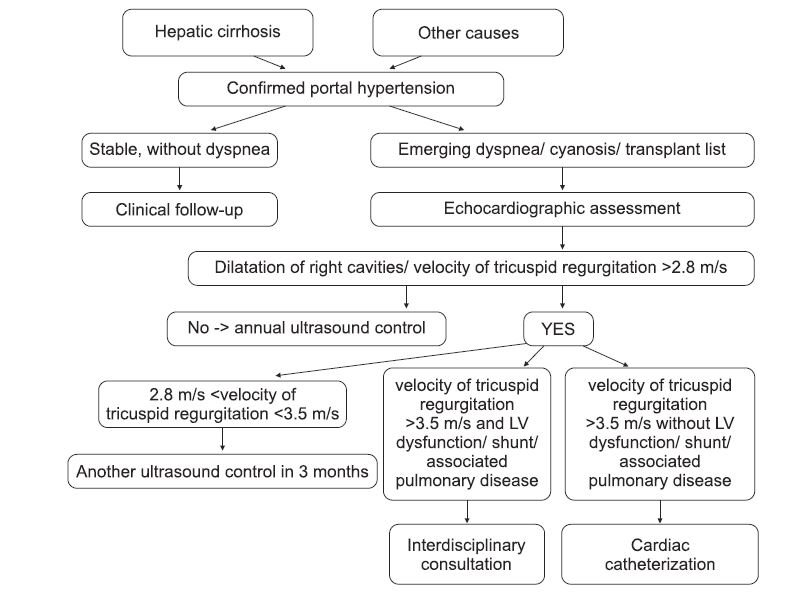
Figure 1. Algorithm for the diagnosis of pulmonary hypertension in patients with confi rmed portal hypertension. Vel = velocity; regurg = regurgitation; tricusp = tricuspid.
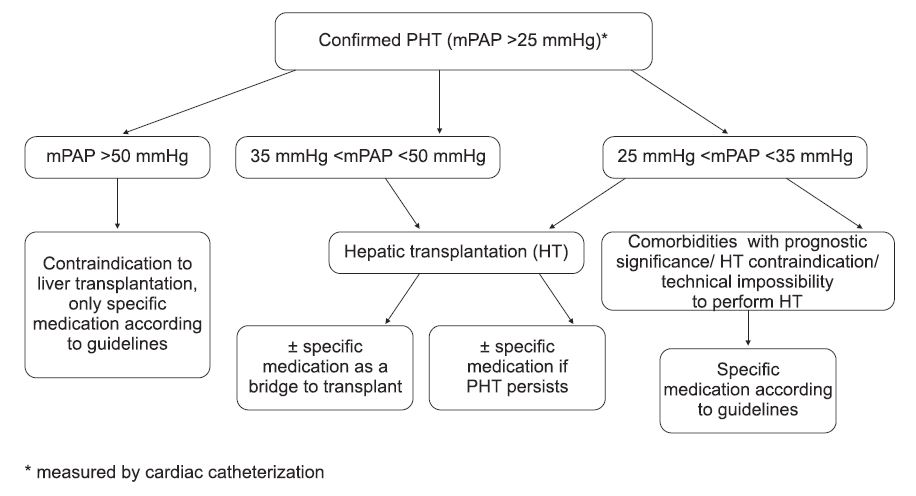
Figure 2. Treatment algorithm proposed for patients with PPHT confi rmed by cardiac catheterization. mPAP = mean pulmonary artery pressure; HT = hepatic transplantation.
ANNEX 1
Table 1. Diagnostic criteria for portal hypertension
| Clinical diagnosis |
| -Collateral circulation -Splenomegaly -Ascites -Edema |
| Non-invasive diagnostic imaging |
| Abdominal ultrasound – portal vein caliber >15 mm – splenic vein caliber >10 mm – highlighting the collateral circulation – highlighting the presents of shunts – increase of intraparenchymal hepatic, spleen, kidney arterial impedance – decrease of mesenteric arterial impedance Upper digestive endoscopy – assesses the complications of portal hypertension (the presence of esophageal, gastric varices, portal-hypertensive gastropathy) |
| Invasive diagnosis |
| Measuring the hepatic venous pressure gradient (HVPG) – gold standard – value >10 mmHg – suggestive of significant portal hypertension |
ANNEX 2
Table 2. Ultrasound parameters suggesting PHT
| Assessment of pulmonary hypertension Assessment of right ventricular (RV) function |
|
PAPs = 4 V² max RT+ RAP (V max RT >2.8 m/s = probable PHT) TAPSE (<16 mm = RV dysfunction) PAPm = 4 V² RP protodiastolic +RAP Percentage variation of VD- FAC area (<35% = RV dysfunction) mPAP = 79-0.45 x TAP (TAP <100 ms- suggestive of PHT) Myocardial performance index – Tei index (>0.4 = RV dysfunction) Dilatation of right cavities (basal RV dimension >42 mm) Right ventricular hypertrophy Pulmonary artery dilatation Excentricity index |
| PAPs = systolic pressure in the pulmonary artery; mPAP = mean pulmonary artery pressure; RAP = right atrial pressure; RT = tricuspid regurgitation; RP = pulmonary regurgitation; SIV= interventricular septum; TAP = pulmonary ascent time; TAPSE = systolic movement of the tricuspid annulus plane; V= velocity; LF = left ventricle; RV = right ventricle. |
Table 3. Estimated right atrial pressure based on the diameter of the inferior vena cava (IVC)
| RAP | 0-5 mmHg | 10-20 mmHg | |
| ≤2.1 cm | ≤2.1 cm | >2.1 cm | >2.1 cm |
| >50% | <50% | >50% | <50% |
* This document represents the opinion of specialists in cardiology, gastroenterology, internal medicine and pulmonology on the topic of common interest of portopulmonary hypertension and it was drawn up following the discussions in the conference held in „Casa Inimii” Sibiu (Heart’s House in Sibiu) (6th-7th February 2015). Participants also agreed on the following 10 practical proposals:
- Correct and constant information of gastroenterology and internal medicine professionals on the prevalence and prognostic impact of portopulmonary hypertension in local/ regional and national
meetings; - Publishing the consensus document on portopulmonary hypertension in national scientific magazines specialized in gastroenterology/ internal medicine / pulmonology/ cardiology;
- Opening a dedicated page on the website of the European Centre for the Study of PHT (PHADER);
- Informing the Romanian Ministry of Health and the Romanian National Health Insurance House on the consensus document and the likely number of patients with portopulmonary hypertension
that could benefit from specific pulmonary vasodilator treatment; - Proposing to the Romanian Ministry of Health and the Romanian National Health Insurance House to include this etiology of PHT in the National Program of Rare Diseases – Pulmonary Hypertension
section; - Creation in the main university centers of interdisciplinary expertise nuclei (gastroenterology, internal medicine, pulmonology, cardiology) for the discussion of the identified cases;
- Promoting the commencement of grants/ doctorates/ bachelor’s degree diplomas on related PPHT subjects;
- Conducting at a time interval of maximum 2 years of a national symposium dedicated to this theme;
- Compulsory input of these patients’ data in the Romanian National Register of Pulmonary Hypertension, thus creating a database useful in clinical research;
- Informing those concerned about the existence of a National Association of Patients with PHT.
References
1. Kuo PC, Plotkin JS, Gaine S, et al. Portopulmonary hypertension and the liver transplant candidate. Transplantation 1999; 67:1087- 1093.
2. Kawut SM, Krowka MJ, Trotter JF, Roberts KE, Benza RL, Badesch DB, et al. Clinical risk factors for portopulmonary hypertension. Hepatology. 2008;48:196–203.
3. Krowka MJ, Plevak DJ, Findlay JY, et al. Pulmonary hemodynamics and perioperative cardiopulmonary- related mortality in patients with portopulmonary hypertension undergoing liver transplantation. Liver Transpl 2000; 6:443- 450.
4. Krowka MJ, Miller DP, Barst RJ, et al. Portopulmonary hypertension: a report from the U.S. –based REVEAL registry. Chest 2012; 141:906- 15
5. Humbert M, Sitbon O, Chaouat A, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med. May 1 2006;173(9):1023-30.
6. Talwalkar JA, Swanson KL, Kwowka MJ, et al. Prevalence of spontaneuous portosystemic shunts in patients with portopulmonary hypertension and effect on treatment. Gastroenterelogy 2011; 141: 1673-1679.
7. Aldenkortt F. et al, Challenges for liver transplantation anesthesia, World J Gastroenterol 2014.
8. MancusoL, Scodato F, Pieri M, et al. Management of portopulmonary hypertension: New Perspectives. World J Gastroenterol 2013; 19: 8252-8257
9. Kennedy JW, Baxley WA, Bunnel IL, et al. Mortality related to cardiac catheterization and angiography. Cathet Cardiovasc Diagn. 1982; 8(4):323-40.
10. Simonneau G, Gatzoulis M., Adatia I. et al, Updated Clinical Classification of Pulmonary Hypertension, JACC 2013; 25:D34-41.
11. Cartin-Ceba R, Swanson K, Iyer V, et al. Safety and efficacy of ambrisentan for the treatment of portopulmonary hypertension. Chest. 2011;139:109–14.
12. Savale L, Magnier R, Le Pavec J, et al. Eur Respir J 2013; 41:96.
13. Reichenberger F, Voswinckel R, Ghofrani HA et al. Eur Respir J. 2006; 28:563
14. Awdish RL, Cajigas HR. Early initiation of prostacyclin in portopulmonary hypertension: 10 years of a transplant center experience. Lung 2013; 191: 593- 600.
15. Starkel P, Vera A, Gunson B, et al. Otcome of liver transplantation for patients with pulmonary hypertension. Liver Transpl 2002; 8: 382-388.
 This work is licensed under a
This work is licensed under a