Ioan Maniţiu1, Elkahlout Ayman2, Radu Cojan3, Cornel Ioan Bitea3, Georgiana Bălţat3
1 Professor of cardiology, Faculty of Medicine, Lucian Blaga University Sibiu, Sibiu Emergency Clinical Hospital, Cardiology I Department
2 Head of Catheterisation Laboratory, Târgu Mureş Emergency Institute of Cardiovascular Disease and Transplant
3 Sibiu Emergency Clinical Hospital, Cardiology I Department
INTRODUCTION
Anomalous origin of the left coronary artery from the pulmonary artery (ALCAPA) is a rare congenital cardiac anomaly with an incidence of 1/300.000 live births (0.4% of all congenital heart disease). ALCAPA is the most common anomaly involving a coronary artery arising from the pulmonary trunk 1,2. The ALCAPA anomaly may result from abnormal septation of the conotruncus into the aorta and pulmonary artery, or from persistence of the pulmonary buds togheter with involution of the aortic buds that eventually form the coronary arteries5. There are two types of ALCAPA syndrome: the infant type and the adult type. Infants experience myocardial infarction and congestive heart failure, and approximately 90% die within the first year of life2-4. In a minority of cases (approximately 20%), sufficient myocardial collaterals develop from the normally arising right coronary artery, and survival into adulthood may occur1; in these situation a left to right shunt into the pulmonary artery is created, leading to a coronary steal phenomenon that results in myocardial ischemia later in life. Few patients survive past childhood without surgical repair, and up to 90% die suddenly at a mean of age of 35 years2. Here we describe a case of adult type ALCAPA syndrom.
CASE REPORT
A 22-year-old woman presents with exertional dyspneea on mild effort from approximately 6 months without any other symptoms. There were no pathological findings at clinical examination, except tachycardia on cardiac auscultation.The patient history prior to current observation include the diagnosis of endocardial fibroelastosis and dilated cardiomyopathy since 5-months-age. At the age of 5 months the patient was admitted to pediatric intensive care unit with severely impaired general condition presenting dyspnea with tachypnea, cough, fever, poor feeding and irritability especially during feeding. The clinical examination at that time revealed grade I dystrophy, respiratory failure signs (tachypnea, sharp pulling in of the chest below and between the ribs with each breath, flaring of the nostrils) and cardiac failure signs (tachycardia, poor peripheral perfusion with cyanosis, dilated jugulars, hepatomegaly with liver at 3 cm below the right costal margin, crackles heard in the lung bases). Cardiac auscultation revealed a II/VI ejection systolic murmur at left sternal border. Chest X ray showed global cardiomegaly, electrocardiogram (ECG) revealed sinus rhythm and deep Q waves in leads DI, aVL, V3-V6. Transthoracic echocardiogram showed dilated left ventricle with poor function and dyskinesia of the apex and the left anterior wall. The pathological signs revealed by clinical examination, chest X ray, ECG, transthoracic echocardiography suggested the diagnosis of a possible anomaly of the coronary system; thus the patient was referred to a more specialized center where she underwent cardiac catheterization and nonselective coronary angiography. Cardiac catheterization showed dilated left ventricle with global hipokinesya and severe systolic dysfunction with an ejection fraction (EF) ~26%, small apical lacunar images (possible mural thrombi or endocardial fibroelastosis), mitral regurgitation grade I, tricuspid regurgitation grade I, slight pulmonary hypertension. Nonselective coronary angiography showed both coronary artery apparently in normal limits. At that time the case was interpreted as endocardial fibroelastosis. Until the age of 11 she received positive inotrope agent and coronarydilator agents, when she stopped this treatment because she was symptom free from the age of 8.
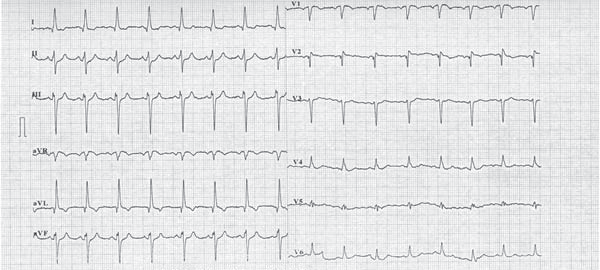
Figure 1. Twelve leads ECG showing sinus tachichardia, left axis deviation, anterosuperior block, q and T wave invertion in DI and aVL leads, Q wave in V1-V2 leads (aspect suggesting anterior myocardial infarction sequel), late transition of QRS complex, microvolt in precordial leads.
Until the age of 22, when she was referred to our unit, she was oligosymptomatic accusing only exertional dyspnea on mild efforts. The ECG on current presentation showed sinus tachycardia with left axis deviation, an anterosuperior block, q waves and T waves inversion in leads DI and aVL, and Q waves in leads V1-V2 (aspect suggesting anterior myocardial infarction sequel), late transition of QRS complex, microvolt in precordial leads (Figure 1). Transthoracic echocardiogram showed a mild left ventricle enlargement with apical dyskinesia, with preserved left ventricle contractile function and an global ejection fraction (measured by Simpson method) of 60% (Figure 2). 2D-Echocardiography revealed also multiple turbulent color flow regions (intracoronary collaterals) in ventricular septum (Figure 3) and was unable to visualize the left coronary artery origin. In the light of the ECG and 2D-Echocardiography findings a coronary CT was obtained and revealed a left main coronary artery with interarterial course, without showing the exact left main coronary artery origin. At this moment the patient was referred to coronary angiography that showed an enlarged right coronary artery (Figure 4) with many collateral branches draining into the pulmonary artery via the left coronary artery (Figure 5, Figure 6). Coronary angiogram confirmed AL-CAPA syndrome and surgical correction was planned. Before surgery a cardiac magnetic resonance imaging (MRI) was made and no signs of scar fibrosis or infarction were found.
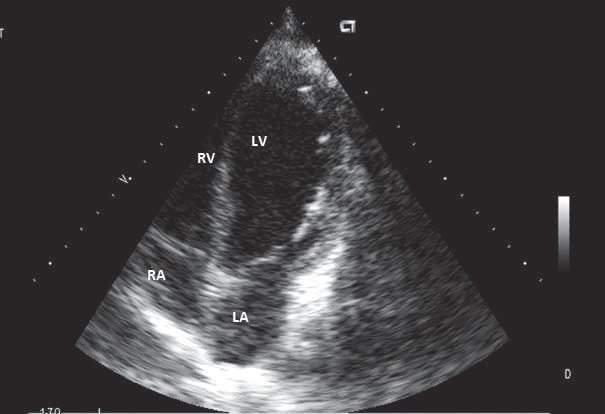
Figure 2. 2D-Echocardiography, 4 chambers view – mild left ventricle anlargement with apical dyskinesia an preserved ejection fraction. LV – left ventricle. RV- right ventricle. LA – left atrium. RA – right atrium.
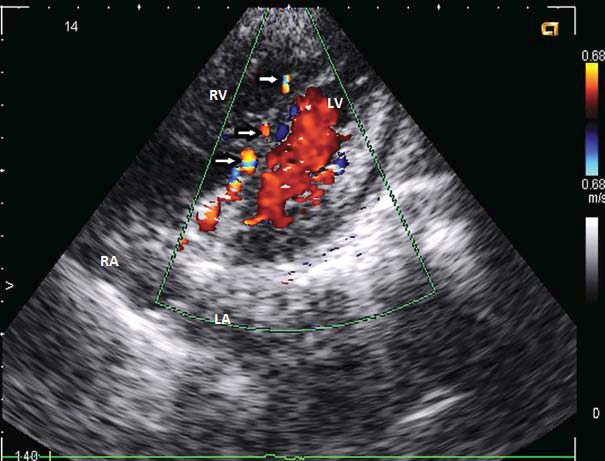
Figure 3. 2D-Echocardiography, 4 chambers view – multiple turbulent color flow regions (intercoronary collaterals) in ventricular septum (white arrows). LV – left ventricle. RV – right ventricle. LA – left atrium. RA – right atrium.
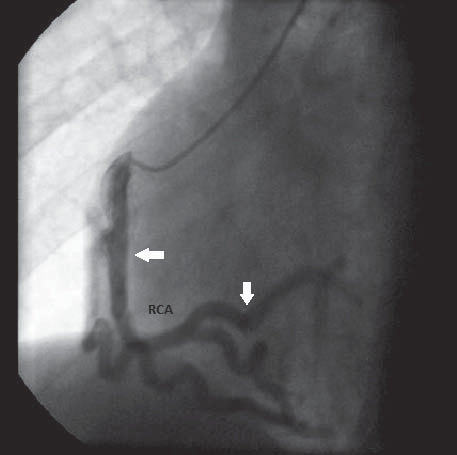
Figure 4. Coronary angiography – catheter insereted intro the right coronary artery ostium; enlarged right coronary artery, without atherosclerosis lesions. RCA – right coronary artery.
DISCUSSIONS
The anomaluos origin of left coronary artery from pulmonary trunk is a well known, althuogh rare, congenital anomaly in humans. In most cases, it’s an isolated anomaly but has occasionaly been associated with other congenital heart defects such as patent ductus arteriosus, ventricular septal defect, tetralogy of Fallot and coarctation of tha aorta6. Our patient presented ALCAPA anomaly without any other congenital heart defects.
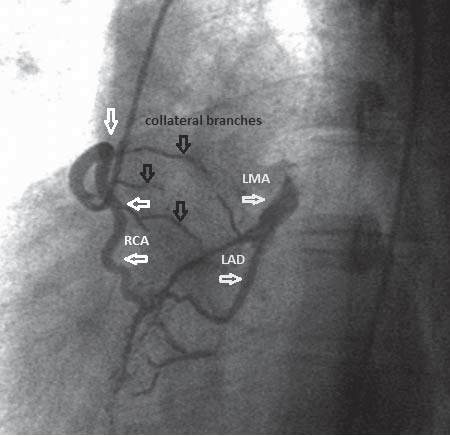
Figure 5.Right coronary angiography in LAO position showing filling of left coronary system through collateral branches (black arrows) originated from right coronary artery. RCA – right coronary artery. LMA – left main coronary artery. LAD – left anterior descending artery.
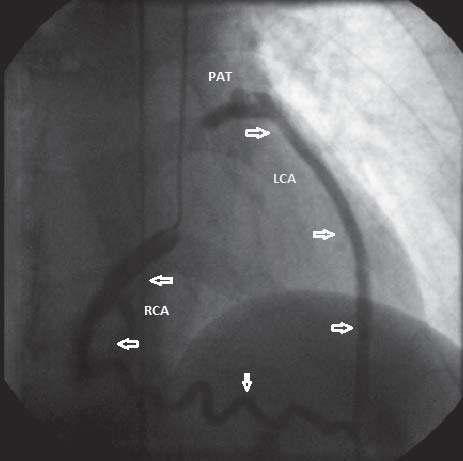
Figure 6. Coronary angiography in RAO position – catheter inserted into right coronary artery ostium; dilated right coronary artery and an abnormal origin of left coronary artery from pulmonary arterial trunk in delayed sequence. RCA – right coronary artery. LCA – left coronary artery. PAT – pulmonary arterial trunk.
The clinical expresion of syndrome results from evolving morphological-functional alterations in pulmonary circulation that occur after birth. Soon after birth, resistance of the pulmonary circulation is so high permitting antegrade flow from the pulmonary artery to left coronary artery, witch perfuses the leftventricle. Therefore, occurence of sudden death is extremely rare in this age group. As pulmonary vascular resistance falls in following weeks, flow from pulmonary artery lo left coronary artery stops and left ventricular perfusion totally depends upon collaterals to left coranary artery developed from right coronary artery. Death ensues if collaterals are poorly developed while on the other hand if colateralls enlarge after an initial period of decompensation, improvement and survival into adulthood occurs – so called adult type of ALCAPA7. In our case there was an initial cardio-respiratory decompensation at 5 months age, when probably the collaterals from right coronary artery to left coronary artery enlarged causing a progressively increase in left coronary flow and a slow remission of clinical signs. In patients who survive into adulthood, pulmonary circulation acts as low resistance siphon and colaterals flow uses left coranary artery as a only conduit into pulmonary circulation thus bypassing the left ventricular myocardium. This coronary steal may cause over tischaemia as well as left ventricular diastolic overload from left to right shunting3,7. In our case, the patient was nearly asymptomatic till date. The presenting symptoms of exertional dyspnea on mild efforts results from myocardial ischaemia due to failure in collateral circulation to ensure a higher flow, and thus more oxygen, to myocardium during effort. Twelve lead ECG can alert the posibility of ALCAPA if ischemic changes are seen especially in young age group. Broad deep Q waves and associated T wave inversion in leads DI and aVL has been described as beeing characteristic for ALCAPA6. Our patient ECG at 5 months, showed deep Q waves in leads DI and aVL, witch is consistent with literature date, and also deep Q waves in leads V3-V6. On ECG signs of infarction are absent in adults7, witch is not consistent with our findings as electrocardiography at 22 years old showed small q waves with T wave inversion in leads DI, aVL and also deep Q waves in leads V1-V2, aspect suggesting anterior myocardial infarction sequel. Echocardiography is an important diagnostic tool for the diagnosis of ALCAPA. In infants with suspicion of endocardial fibroelastosis or dilated cardiomyopathy, anomalous origin of coronary artery must be ruled out6. Our case was first interpreted as endocardial fibroelastosis based on radiographic (cardiomegaly), echochardiographic (dilated left ventricle with reduced ejection fraction) and angiocoronarographic (nonselective coronary angiography showed both coronary artery apperently in normal limits) findings at 5 months age. This misinterpretation was principally caused by the cronary angiography that showed both coronary artery apperently normal; we must specify that the pacient underwent a nonselective coronary angiography that has its limits. In ALCAPA pacients surviving beyond one year without treatment, coronary collaterals have been found to be obvious septal color flow signals echocardiographically. The identification of these septal collaterals is the initial clue for the diagnosis of ALCAPA in patients over one year of age6. This echocardiographic aspect was found in our pacient at 22 years old; this finding rised the suspicion of a coronary system anomaly. Also, 2D- echocardiography did not revealed mitral regurgitation, witch is a common finding in ALCAPA patients. Because the ostium of the left coronary artery was not visualized the patient was submited to other investigations to confirm the diagnosis. Additional imaging techniques such as CT scan/MRI are undertaken only when definitive diagnosis by echocardiography is not possible, or in an effort to exclude other potential diagnoses 6. In our case coronary CT revealed a left main coronary artery with interarterial course, without showing the exact left main coronary artery origin; therefore we performed selective coronary angiography to confirm the diagnosis. Three angiographic criteria are establised for diagnosing ALCAPA. They are as follows7: 1) retrograde filling of left coronary artery, 2) connection of left coronary artery with pulmonary artery and 3) absence of left coronary artery originating from aorta. Since all three criteria are fulfilled in our case, diagnosis of ALCAPA is confirmed. The adult form of ALCAPA is characterised by exuberance in collateral coronary circulation, witch allows survival until adulthood, with case beeing reported at the age of 72 years7. When coronary artery anomalies like ALCAPA are diagnosed, urgent surgery is often indicated in order to prevent myocardial ischemia, malignant ventricular arrhythmias and sudden cardiac death 8. In cases of ALCAPA several surgical treatment options have been proposed9. Today surgical procedures are aimed at creating a two-coron ary system either via 1) a bypass graft (mammary artery or saphenous vein) in combination with ligation of the anomalous artery, 2) the Takeuchi-procedure where an intrapulmonary tunnel from the aortopulmonary window to the coronary artery is created or 3) translocation of the left coronary artery from the pulmonary trunk to the aortic sinus10,11. The latter depends on the distance between the origin o f the anomalous artery and the aorta, but is possible in the majority of the cases12. In our case, the pacient was proposed for bypass graft with mamary artery. In infants, most of the patients with corrected ALCAPA show normalization of both ventricular function and mitral valve insufficiency13,14. Estimated long-term survival at 20 years was recently s hown to be 94.8%14. No long-term studies of large populations of adults with corrected ALCAPA are available, but the prognosis is generally good. The late outcome after revascularization mainly depends on the extend of irreversible impaired left ventricular function and the presence of myocardial scar tissue15. In our case cardiac MRI was made and no signs of scar fibrosis or infarction were found. Restoration of a dual coronary system will prevent further ischemia and arrhythmias of acute ischemic origin, but the anatomical substrate for ventricular arrhythmias in patients with old myocardial infarction will not be altered after revascularization. Treatment options include drug therapy, implantable cardiac device (ICD) implantation or catheter ablation. ICD implantation has been shown to be superior to drug treatment in patients with a history of ventricular tachycardia/ventricular fibrillation and previous myocardial infaction (secondary prophylaxis) – especially when there is left ventricular dysfunction15. With recurrent ventricular tachycardia refractory to these treatment options, direct surgical ablation or resection of the arrhythmogenic focus is an option12. ICD implantation may also be considered in patients without a history of arrhythmias, when there is marked left ventricular dysfunction and electrophysiological study shows inducible ventricular tachycardia (primary prophylaxis) 12. In our patient antiarrhythmic treatment is controversial co nsidering that no arrhythmia was documented in her history, left ventricle sistolic function is preserved and cardiac MRI found no signs of scar fibrosis. In this situation is required long-term clinical and electrocardiographic (ECG and Holter ECG) monitoring.At this moment there are no clear indications regarding pregnancy in ALCAPA womens. Pregnancy is associated with cardiac overload and may be a trigger for cardiac decompensation in these patients. There are case reports in witch some patients presented cardiac decompensation during pregnancy, but in others, ALCAPA syndrome was diagnosed after some period after birth.
CONCLUSION
Diagnosis of adulthood ALCAPA should be considered not only in adult patients presenting with clear evidence of ischemic heart disease, left ventricular dysfunction, or arrhythmias, but also more importantly in patients with minor symptoms of exercise intolerance or dyspnea that could be easily misinterpreted. ALCAPA syndrome is a congenital anomaly which must be suspected in infants and adults in the presence of ischemic electrocardiographic changes. In adults 2D-echocardiography is a very useful diagnostic tool. Additional imaging investigations are reserved for cases with uncertain echocardiographic diagnosis. Pacients prognosis depends on the severity of left ventricular dysfunction and the risk of malignant arrhythmias. ALCAPA syndrome treatment is essentially surgical and should be done as soon as the diagnosis is established. Antiarrhythmic therapy should be individualized according to each patient. Conflict of interest: none declared.
List of abbreviations:
ALCAPA Anomalous Left Coronary Artery from Pulmonary Artery
ECG electrocardiogram
2D- Echocardography 2 dimentional- Echocardography
EF ejection fraction
coronary CT coronary computed tomography
MRI magnetic resonance imaging
ICD implantable cardiac device
References
1. Taylor AJ, Virmani R. Coronary artery anomalies.In Cardiology, Third edition. Eds: MH Crawford, JP DiMarco, WJ Paulus et al. Eslsevier, Philadelphia, 2010, 231-241.
2. Ghaderi F, Gholoobi A, Moeinipour A. Unique Echocardiographic Markers of Anomalous Origin of the Left Coronary Artery from the Pulmonary Artery (ALCAPA) in the Adult. Echocardiography, 2014;
31:E13–E15.
3. Rugina AL, Iordache C, Jitareanu C, Mihaila D. Sindromul Bland- White-Garland cu evolutie fatala precoce. Revista Romana de Pediatrie, 2012; 3:317-322.
4. Oprea A, Popa A, Radulescu B, Postu M, Musteata M, Ginghina C. Originea anormala a arterei coronare stangi din artera pulmonara. Stetoscop, 2008; 76:28-33.
5. Liu Y, Miller BW. ALCAPA Presents in an Adult with Exercise Intolerance but Preserved Cardiac Function. Hindawi Publishing Corporation, Case Reports in Cardiology, 2012; 2012: Article ID 471759, 3 pages.
6. Dilawar M, Ahmad Z. Anomalous left coronary artery from pulmonary artery: Case series and brief review. Journal of Pediatrics, 2012; 2:77-81.
7. Parale GP, Pawar SS. Adult Type Anomalous Left Coronary Artery fromPulmonary Artery. Journal of the Association of Physicians of India, 2006; 54:397-399.
8. Lange R, Vogt M, Hörer J, Cleuziou J, Menzel A, Holper K, Hess J, Schreiber C. Long-term results of repair of anomalous origin of the left coronary artery from the pulmonary artery. Annals of Thoracic Surgery, 2007; 83:1463-1471.
9. Collins N, Colman J, Benson L, Hansen M, Merchant N, Horlick E. Successful percutaneous treatment of anomalous left coronary artery from pulmonary artery. International Journal of Cardiology,
2007;122:e29-e31.
10. Dahle G, Fiane AE, Lindberg HL. ALCAPA, a possible reason for mitral insufficiency and heart failure in young patients. Scandinavian Cardiovascular Journal, 2007; 41:51-58.
11. Michielon G, Di CD, Brancaccio G, Guccione P, Mazzera E, Toscano A, Di Donato RM. Anomalous coronary artery origin from the pulmonary artery: correlation between surgical timing and left ventricular function recovery. Annals of Thoracic Surgery, 2003; 76:581-588.
12. Kristensen T, Kofoed KF, Helqvist S, Helvind M, Søndergaard L. Anomalous origin of the left coronary artery from the pulmonary artery (ALCAPA) presenting with ventricular fi brillation in an adult: a case report. Journal of Cardiothoracic Surgery, 2008; 3:33-38.
13. Isomatsu Y, Imai Y, Shin’oka T, Aoki M, Iwata Y. Surgical intervention for anomalous origin of the left coronary artery from the pulmonary artery: the Tokyo experience. Journal of Thoracic and Cardiovascular Surgery, 2001; 121:792-797.
14. Lange R, Vogt M, Horer J, Cleuziou J, Menzel A, Holper K, Hess J, Schreiber C. Long-term results of repair of anomalous origin of the left coronary artery from the pulmonary artery. Annals of Thoracic Surgery, 2007; 83:1463-1471.
15. Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, Gregoratos G, Klein G, Moss AJ, Myerburg RJ, Priori SG, Quinones MA, Roden DM, Silka MJ, Tracy C, Smith SC Jr., Jacobs AK,
Adams CD, Antman EM, Anderson JL, Hunt SA, Halperin JL, Nishimura R, Ornato JP, Page RL, Riegel B, Blanc JJ, Budaj A, Dean V, Deckers JW, Despres C, Dickstein K, Lekakis J, McGregor K, Metra
M, Morais J, Osterspey A, Tamargo JL, Zamorano JL. ACC/AHA/ESC 2006 Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: a report
of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop Guidelines for
Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death): developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation, 2006; 114:e385-e484.
 This work is licensed under a
This work is licensed under a